分子動力学シミュレーション解析であるMM-GBSA/PBSA、decomposition、アラニンスキャン解析を論文に沿ってやってみた。
本記事はリガンド-タンパク質 MDsimulation(分子動力学シミュレーション)を論文に沿って行なった結果に対し、
論文で使われている解析方法であるMM-GBSA解析とMM-PBSA解析、decomposition解析、アラニンスキャン解析を行なっています。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB
本記事は以下の論文のフォローになります。
※本論文のMD simulationはAMBERを用いて行なっておりますが、本記事ではGROMACSでフォローしています。
本論文は標的の選定方法からスクリーニング、スクリーニングした薬物候補化合物の毒性、物性予測、MDシミュレーションまでしており、ほぼ無料のツールですることができます。in silico創薬の基本が詰まっているので、一緒に勉強していきましょう。
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
In silico創薬とその流れ
In silico 創薬、in silicoスクリーニングとは
インシリコ創薬(in silico drug discovery)は、コンピュータを駆使し、シミュレーションやデータ解析を用いて新薬を設計・発見する手法です。
これにより、従来の実験的な方法に比べてコストと時間を大幅に削減できるとされています。
その中でもin silicoスクリーニングは、膨大なデータベースから有望な候補物質を迅速に特定し、その効果や副作用を予測することで、創薬の初期段階での効率化が図られます。
一見難しそうなin silico創薬ですが、現在では様々なアプリケーションやwebサイトがあり、それらを駆使すれば、誰でも簡単に創薬をすることもできます。本記事では、それらのアプリケーションを駆使し、in silico創薬を行った論文をもとに、手法をわかりやすく説明していきます。
In silico創薬の流れ
- ターゲット選定、準備:疾患の原因となる分子(ターゲット)を特定し、その3D構造を準備します。
- 化合物ライブラリの構築:in silicoスクリーニングに使用する化合物集団(=ライブラリ)を作成します。
- in silicoスクリーニング:コンピュータ上で数百万の化合物を対象に、ターゲット分子との結合親和性を評価します。
-
分子動力学シミュレーション(MD simulation):候補化合物とターゲットの相互作用を動的に解析し、安定性や効果を予測します。
4.1.環境構築:Windowsでの環境構築を示しています。
4.2.MD simulation:MD simulationを行います。結果の可視化を行います。
4.3.RMSD, RMSF, RoG, hydrogen bond, RDF 解析:構造変化や分子間相互作用を評価するための解析手法を行います。
4.4.MM-GBSA解析とMM-PBSA解析、decomposition解析、アラニンスキャン(本記事):分子間相互作用や結合エネルギーへの各残基の寄与を解析します。 - 物性、毒性評価:過去のデータを用いて、新たな候補物質の特性や副作用を予測するモデルを構築します。
- (実験的検証):in silico解析で得られた候補物質を実験室で合成し、実際の効果や安全性を検証します。
すでに公開したものについては、リンクを貼っています。今回はMaterial and Methodの以下の部分をフォローしています。
本章ではMM-GBSA解析とMM-PBSA解析、decomposition解析、アラニンスキャンを一気に行います。対応するFigure, Tableは以下の通りです。
-
Table1(MM-GBSA解析とMM-PBSA解析)
ここではMM-GBSA解析とMM-PBSA解析を行っています。
MM-GBSA解析とMM-PBSA解析はどちらも分子の結合自由エネルギーを計算する方法ですが、溶媒効果の扱い方が異なります。以下、簡単にその違いを説明します。
MM-GBSA (Molecular Mechanics - Generalized Born Surface Area)
-
溶媒モデル: GBモデル(Generalized Born model) を使用します。
- 溶媒の影響を計算するときに、分子の周りにある水分子の効果を近似的に取り入れる方法です。
- GBモデルは計算が比較的軽く、迅速に解析を行えるため、大規模な計算や多くのサンプルに適しています。
- 用途: 大量の構造を高速に解析する際に便利ですが、溶媒効果の精度はPBSAに比べると劣る場合があります。
MM-PBSA (Molecular Mechanics - Poisson-Boltzmann Surface Area)
-
溶媒モデル: PBモデル(Poisson-Boltzmann model) を使用します。
- 分子周囲の溶媒効果をより詳細に計算できるモデルで、電荷分布をPoisson-Boltzmann方程式で扱います。
- より正確な溶媒の影響を得られるため、精度が必要な解析に適していますが、計算が重くなることが多いです。
- 用途: 1つのリガンド-タンパク質複合体に対する詳細な解析や、特に水の影響が重要な場合に使用されます。
-
溶媒モデル: GBモデル(Generalized Born model) を使用します。
-
Figure 8 (decomposition解析)
MM-GBSAやMM-PBSA解析のdecomposition(エネルギー分解解析)は、酵素とリガンド複合体の結合自由エネルギーを各残基に分解し、ホットスポット残基を特定する方法です。結合エネルギーが1 kcal/mol以下の残基は、リガンドとの結合に重要とされ、例えばHis398、Arg399、Ser238、Asp319などがホットスポットとして挙げられます。この解析により、創薬や分子設計において特に重要な結合部位を把握し、アラニンスキャンなどで結合強度への影響を評価するための基礎情報が得られます。
-
Table2 アラニンスキャン
アラニンスキャン(Alanine Scanning)は、タンパク質の特定の残基をアラニン(ALA)に置き換え、その変異がタンパク質の機能やリガンド結合に与える影響を評価する方法です。この手法は、タンパク質内の特定残基が結合や活性にどの程度寄与しているかを調べるのに用いられます。
本記事で用いられるツール一覧
gmx_MMPBSA:MM-GBSA解析とMM-PBSA解析、decomposition解析、アラニンスキャンを一気に行うことのできるツールです。
gmx_MMPBSAの環境構築
まずは環境構築です。
env.ymlのファイルを作成します。
テキストファイルを開き、以下をコピペして、env.ymlとして、作成してください。
gmx_MMPBSAのホームページに従って、インストールできますが、
このリンクによると、python=3.9でないといけないです!そのため、pythonのバージョンは変更してください!
(作成したファイルはどこに移動させてもよいのですが、MMPBSA_GBSAなどと新しくディレクトリを作って、そこに入れるとよいでしょう。)
name: gmxMMPBSA
channels:
- conda-forge
- defaults
dependencies:
- python=3.9
- ambertools<=23.3
- mpi4py<=3.1.5
- gromacs<=2023.4
- git
- pyqt=5.15.4
- pandas=1.2.2 # Install via Conda instead of pip
- seaborn<0.12
- scipy>=1.6.1
- matplotlib=3.5.2
- tqdm
- pip
- pip:
- gmx-mmpbsa # Only keep what is not available via Conda
続いて、env.ymlファイルがあるディレクトリに行き、以下を実行します、仮想環境を作ります。
# Create a new environment and use the *.yml file to install dependencies
conda env create -n gmxMMPBSA --file env.yml
# To use gmx_MMPBSA, just activate the environment
conda activate gmxMMPBSA
conda env create -n gmxMMPBSA --file env.yml
仮想環境を作成して、*.yml ファイルから依存関係をインストールする
-
conda env create: 新しい仮想環境を作成するコマンドです。 -
n gmxMMPBSA: このオプションは、作成する仮想環境の名前を指定します。この場合、環境の名前はgmxMMPBSAです。 -
-file env.yml: このオプションは、依存関係(パッケージ)を記述した.ymlファイルを指定します。このenv.ymlファイルには、Python のバージョンや必要なパッケージのリストが書かれています。condaは、このファイルを使って環境に必要なパッケージをインストールします。
conda activate gmxMMPBSA
作成した仮想環境をアクティブにする
-
conda activate: このコマンドは、指定した仮想環境をアクティブにします。仮想環境がアクティブになると、その環境でインストールされた特定のバージョンの Python やライブラリ、ツールが使用可能になります。 -
gmxMMPBSA: 作成した仮想環境の名前です。この環境をアクティブにすることで、その環境内のgmx_MMPBSAというツールを使用できるようになります。
次に現在のディレクトリにtestディレクトリを作り、以下を実行してください!
きちんとgmxMMPBSA ができるかテストできます。
gmx_MMPBSA_test -f test -n 10
実行が終了すると、以下の画面になると思います。

これで環境のセットアップは完了です。
Acceptを押してください。
右側のタブを適当に押すと、テストデータのグラフが見れます。
これにて、環境構築は完成です。
いったん閉じて大丈夫です。
解析の実行
以下のコードを実行してください。
gmx_MMPBSA --create_input gb pb ala decomp
コマンド gmx_MMPBSA --create_input gb pb ala decomp は、gmx_MMPBSAで以下の解析を実行するための入力ファイルを作成するオプションを指定しています。それぞれのオプションの意味は次のとおりです。
-
gb:Generalized Born (GB) モデルを使ったMM-GBSA解析用の入力ファイルを作成します。このモデルは溶媒効果を取り入れて結合エネルギーを計算します。 -
pb:Poisson-Boltzmann (PB) モデルを用いたMM-PBSA解析用の入力ファイルを作成します。PBモデルはより詳細な溶媒効果を考慮し、結合エネルギーの精度を高めることができますが、計算量が多くなります。 -
ala:アラニンスキャン(Alanine Scanning) 用の入力ファイルを作成します。特定の残基をアラニンに置換し、結合自由エネルギーの変化を評価します。ホットスポット残基の寄与を詳細に解析するために有用です。 -
decomp:エネルギー分解(Decomposition)解析 用の入力ファイルを作成します。これにより、各残基が結合自由エネルギーにどの程度寄与しているかを計算し、特に重要な残基(ホットスポット)を特定することが可能です。
mmpbsa.inが生成されています。これを開いてください。
以下を変更してください。(他の設定はデフォルトのままです)
sys_name→お好きな名前
endframe→simulaitonしたframe数
forcefields→oldff/leaprc.ff99SBildn (今回はamber99sb-ildnを力場に設定しているため)
mutant_res →"A/710" (A鎖のArg339、一つずつしか無理そうです)
idecomp →”1”(標準的な結合エネルギー解析)
print_res →”within 6”(リガンドに近い6つのアミノ酸)
Input file generated by gmx_MMPBSA (v1.6.3)
Be careful with the variables you modify, some can have severe consequences on the results you obtain.
# General namelist variables
&general
sys_name = "Protein-ligand" # System name
startframe = 1 # First frame to analyze
endframe = 500 # Last frame to analyze
interval = 1 # Number of frames between adjacent frames analyzed
forcefields = "oldff/leaprc.ff99SBildn" # Define the force field to build the Amber topology
ions_parameters = 1 # Define ions parameters to build the Amber topology
PBRadii = 3 # Define PBRadii to build amber topology from GROMACS files
temperature = 298.15 # Temperature
qh_entropy = 0 # Do quasi-harmonic calculation
interaction_entropy = 0 # Do Interaction Entropy calculation
ie_segment = 25 # Trajectory segment to calculate interaction entropy
c2_entropy = 0 # Do C2 Entropy calculation
assign_chainID = 0 # Assign chains ID
exp_ki = 0.0 # Experimental Ki in nM
full_traj = 0 # Print a full traj. AND the thread trajectories
gmx_path = "" # Force to use this path to get GROMACS executable
keep_files = 2 # How many files to keep after successful completion
netcdf = 0 # Use NetCDF intermediate trajectories
solvated_trajectory = 1 # Define if it is necessary to cleanup the trajectories
verbose = 1 # How many energy terms to print in the final output
/
# (AMBER) Generalized-Born namelist variables
&gb
igb = 5 # GB model to use
intdiel = 1.0 # Internal dielectric constant for sander
extdiel = 78.5 # External dielectric constant for sander
saltcon = 0.0 # Salt concentration (M)
surften = 0.0072 # Surface tension
surfoff = 0.0 # Surface tension offset
molsurf = 0 # Use Connelly surface ('molsurf' program)
msoffset = 0.0 # Offset for molsurf calculation
probe = 1.4 # Solvent probe radius for surface area calc
ifqnt = 0 # Use QM on part of the system
qm_theory = "" # Semi-empirical QM theory to use
qm_residues = "" # Residues to treat with QM
com_qmmask = "" # Mask specifying the quantum atoms in complex
rec_qmmask = "" # Mask specifying the quantum atoms in receptor
lig_qmmask = "" # Mask specifying the quantum atoms in ligand
qmcharge_com = 0 # Charge of QM region in complex
qmcharge_lig = 0 # Charge of QM region in ligand
qmcharge_rec = 0 # Charge of QM region in receptor
qmcut = 9999.0 # Cutoff in the QM region
scfconv = 1e-08 # Convergence criteria for the SCF calculation, in kcal/mol
peptide_corr = 0 # Apply MM correction to peptide linkages
writepdb = 1 # Write a PDB file of the selected QM region
verbosity = 0 # Controls the verbosity of QM/MM related output
alpb = 0 # Use Analytical Linearized Poisson-Boltzmann (ALPB)
arad_method = 1 # Selected method to estimate the effective electrostatic size
/
# (AMBER) Possion-Boltzmann namelist variables
&pb
ipb = 2 # Dielectric model for PB
inp = 1 # Nonpolar solvation method
sander_apbs = 0 # Use sander.APBS?
indi = 1.0 # Internal dielectric constant
exdi = 80.0 # External dielectric constant
emem = 4.0 # Membrane dielectric constant
smoothopt = 1 # Set up dielectric values for finite-difference grid edges that are located across the solute/solvent dielectric boundary
istrng = 0.0 # Ionic strength (M)
radiopt = 1 # Use optimized radii?
prbrad = 1.4 # Probe radius
iprob = 2.0 # Mobile ion probe radius (Angstroms) for ion accessible surface used to define the Stern layer
sasopt = 0 # Molecular surface in PB implict model
arcres = 0.25 # The resolution (Å) to compute solvent accessible arcs
memopt = 0 # Use PB optimization for membrane
mprob = 2.7 # Membrane probe radius in Å
mthick = 40.0 # Membrane thickness
mctrdz = 0.0 # Distance to offset membrane in Z direction
poretype = 1 # Use exclusion region for channel proteins
npbopt = 0 # Use NonLinear PB solver?
solvopt = 1 # Select iterative solver
accept = 0.001 # Sets the iteration convergence criterion (relative to the initial residue)
linit = 1000 # Number of SCF iterations
fillratio = 4.0 # Ratio between the longest dimension of the rectangular finite-difference grid and that of the solute
scale = 2.0 # 1/scale = grid spacing for the finite difference solver (default = 1/2 Å)
nbuffer = 0.0 # Sets how far away (in grid units) the boundary of the finite difference grid is away from the solute surface
nfocus = 2 # Electrostatic focusing calculation
fscale = 8 # Set the ratio between the coarse and fine grid spacings in an electrostatic focussing calculation
npbgrid = 1 # Sets how often the finite-difference grid is regenerated
bcopt = 5 # Boundary condition option
eneopt = 2 # Compute electrostatic energy and forces
frcopt = 0 # Output for computing electrostatic forces
scalec = 0 # Option to compute reaction field energy and forces
cutfd = 5.0 # Cutoff for finite-difference interactions
cutnb = 0.0 # Cutoff for nonbonded interations
nsnba = 1 # Sets how often atom-based pairlist is generated
decompopt = 2 # Option to select different decomposition schemes when INP = 2
use_rmin = 1 # The option to set up van der Waals radii
sprob = 0.557 # Solvent probe radius for SASA used to compute the dispersion term
vprob = 1.3 # Solvent probe radius for molecular volume (the volume enclosed by SASA)
rhow_effect = 1.129 # Effective water density used in the non-polar dispersion term calculation
use_sav = 1 # Use molecular volume (the volume enclosed by SASA) for cavity term calculation
cavity_surften = 0.0378 # Surface tension
cavity_offset = -0.5692 # Offset for nonpolar solvation calc
maxsph = 400 # Approximate number of dots to represent the maximum atomic solvent accessible surface
maxarcdot = 1500 # Number of dots used to store arc dots per atom
npbverb = 0 # Option to turn on verbose mode
/
# Alanine scanning namelist variables
&alanine_scanning
mutant_res = "A/710" # Which residue will be mutated
mutant = "ALA" # Defines if Alanine or Glycine scanning will be performed
mutant_only = 0 # Only compute mutant energies
cas_intdiel = 0 # Change the intdiel value based on which aa is mutated
intdiel_nonpolar = 1 # intdiel for nonpolar residues
intdiel_polar = 3 # intdiel for polar residues
intdiel_positive = 5 # intdiel for positive charged residues
intdiel_negative = 5 # intdiel for negative charged residues
/
# Decomposition namelist variables
&decomposition
idecomp = 1 # Which type of decomposition analysis to do
dec_verbose = 0 # Control energy terms are printed to the output
print_res = "within 6" # Which residues to print decomposition data for
csv_format = 1 # Write decomposition data in CSV format
/
次のコードを実行する前に以下のファイルがあることを確認してください。
該当するファイルがないとエラーになり、足りないファイルが示されるので、その都度足していけば大丈夫です。

以下を実行してください。
gmx_MMPBSA -O -i mmpbsa.in -cs md_0_10.tpr -ct md_0_10_center.xtc -ci index.ndx -cg 1 13 -cp topol.top -o MMGBPBSA_ALA_decomp.dat -eo MMGBPBSA_ALA_decomp.csv
-
gmx_MMPBSA: このコマンドはGROMACSとMMPBSAの連携を実現するプログラムを呼び出します。MMPBSAは分子系の結合自由エネルギーを計算するための手法で、特に薬物設計やタンパク質相互作用研究に使われます。 -
O: 出力ファイルを強制的に上書きします。すでに同名のファイルが存在する場合、そのファイルが上書きされます。 -
i mmpbsa.in: 入力ファイルmmpbsa.inは、MM/GBSA計算に関する設定情報を含んでいます。このファイルには、エネルギー計算の詳細なパラメータや、どのエネルギー項を計算するかなどの指示が記述されています。 -
cs md_0_10.tpr: 系の座標とトップロジー情報を含むGROMACSの入力ファイルです。このファイルは分子動力学シミュレーションを開始する際に必要な情報を提供します。 -
ct md_0_10_center.xtc: トラジェクトリファイルです。このファイルには、シミュレーションの座標情報が含まれており、各時点の系の状態を表します。MM/GBSA解析では、この座標情報に基づいてエネルギー計算が行われます。 -
ci index.ndx: インデックスファイルです。このファイルには、分子の特定の部分を定義したグループのリストが含まれています。たとえば、タンパク質の一部やリガンドをグループとして指定することができます。 -
cg 1 13: グループを指定するオプションで、ここでは1番目と13番目のグループが選ばれています。これらのグループ間の相互作用エネルギーを計算します。インデックスファイル内で定義されているグループ番号を使用して、特定の分子や分子群を選択します。 -
cp topol.top: トポロジーファイルtopol.topは、分子の結合情報や力場パラメータを含んでいます。MM/GBSA解析の際に必要な分子の詳細情報を提供します。 -
o MMGBPBSA_ALA_decomp.dat: MM/GBSAエネルギー計算の結果を含む出力ファイルです。このファイルに、アラニンスキャンデコンポジションを含む解析結果が保存されます。 -
eo MMGBPBSA_ALA_decomp.csv: エネルギー分解の結果をCSV形式で保存するための出力ファイルです。このファイルには、各残基がどれだけのエネルギーを寄与しているかが記録され、エクセルなどで分析するのに適しています。
MM-GBSA PBSA解析結果
丸々2日くらいかかります。実行が完了すると、以下のように出力されます。
PB処理に大幅に時間がかかっていることがわかります。

また以下のanalyticsのウィンドウが立ち上がっています。
今回の解析結果はProtein-ligandなので、こちらを選択し、Acceptを押します。

閉じてしまった場合はgmx_MMPBSA_ana とを実行すれば、出てきます。
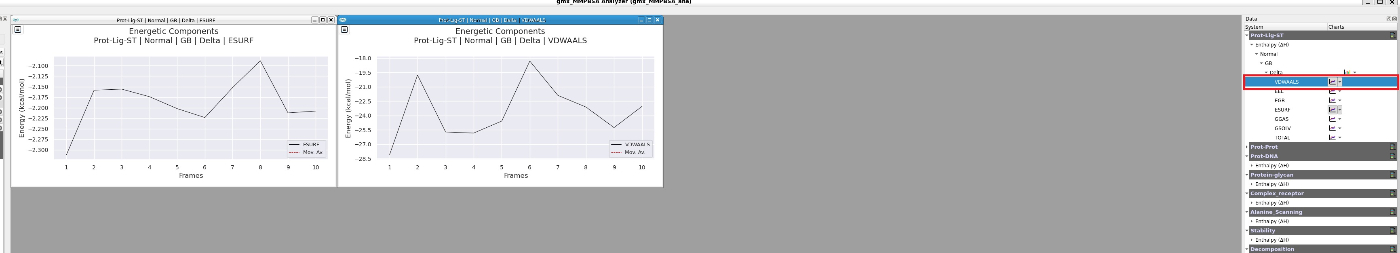
まずはMM-PBSAとMMGBSAの結果を見てみます。
論文のTable1(MM-GBSA解析とMM-PBSA解析)に該当しています。
出力ファイルMMGBPBSA_ALA_decomp.dat が生成しているので、こちらを開いてみてください。
GENERALIZED BORN: と書いてある以下に対応する箇所が該当箇所です。

以下のように対応しています。これを論文の表にすればOKです。
| 画像の項目名 | 提示されたエネルギー成分(ラベル) | 提示された値(例) |
|---|---|---|
| ΔG binding | ΔTOTAL(結合自由エネルギー) | -22.11 |
| ΔG electrostatic | ΔEEL(静電相互作用) | -30.39 |
| ΔG bind van der Waals | ΔVDWAALS(ファンデルワールス相互作用) | -19.96 |
| ΔG bind gas phase | ΔGGAS(ガス相での結合エネルギー) | -50.35 |
| ΔG polar solvation | ΔEGB(極性溶媒和エネルギー) | 31.52 |
| ΔG non polar solvation | ΔESURF(非極性溶媒和エネルギー) | -3.27 |
| ΔG solvation | ΔGSOLV(全体の溶媒和エネルギー) | 28.25 |
同様に
POISSON BOLTZMANN: もあるので、この値を参考にします。

decomposition解析結果
続いて、Figure8のdecomposition解析の結果を表示します。
analysis画面のDecompositionのTDCの棒グラフを開いてみてください。
Figure 8のようなグラフができます。

現状ではリガンドもあるので、虫眼鏡マークを押し、残基だけを拡大するとリガンドだけが表示されます。

-
負のエネルギー値(例:
A:LYS:334やA:LEU:357など): これらの残基は、タンパク質とリガンド間の相互作用を安定化する寄与をしています。負のエネルギー値が大きいほど、結合に対する貢献が高いことを示します。 -
正のエネルギー値(例:
A:ARG:710やA:SER:737): これらの残基は、リガンドとの相互作用に対して不安定化の寄与をしている可能性があります。正のエネルギー値が大きいほど、結合に対して抑制的な影響を及ぼしている可能性があります。 - エラーバー: 各バーの上にあるエラーバーは、不確実性を示しており、各残基のエネルギー寄与のばらつきを表します。エラーバーが長いほど、その値のばらつきが大きく、測定値の信頼性が低くなる可能性があります。
アラニンスキャン解析結果
ここではTable2のようなアラニンスキャンの結果を解析していきます
こちらは以下のDeltaの右側の▼を押し、出てくるShow in TableのAverageのTotalを比べれば、Alanineに変えたことによるエネルギーの変化を見ることができます。

左通常の値 右アラニンスキャンした値
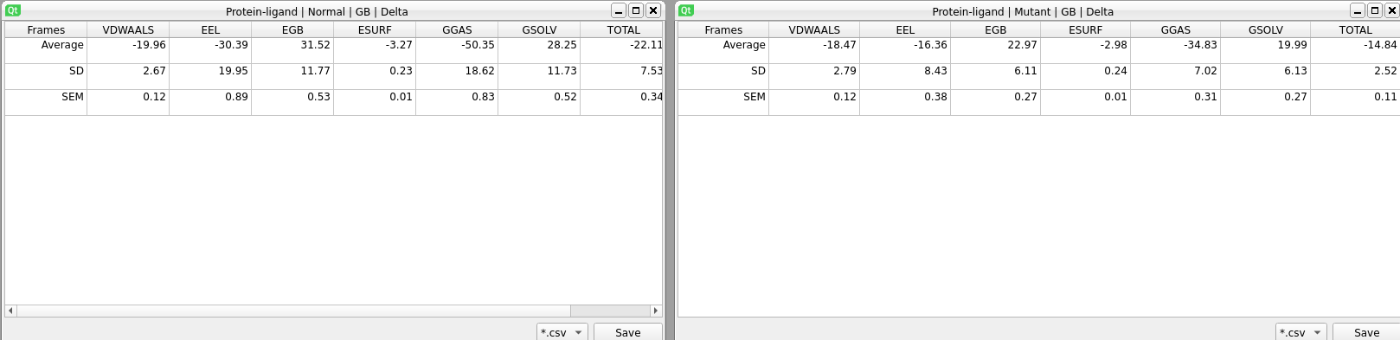
ここでは通常の値(Total)が-22.11, アラニン変異体(Total)が-14.84なので、Arg339が7.27 kcal/molほどのエネルギーに寄与していることがわかります。
参考文献
Discussion