インシリコ創薬の分子ドッキングで必要なタンパク質の準備を論文に沿ってやってみる。【in silico創薬】
本記事ではIn silico創薬の論文をフォローし、標的タンパク質の決定の仕方とその準備について説明しています。この記事の内容が理解できるようになると、標的タンパク質の決定の仕方と分子ドッキングなどに使われるタンパク質を準備することができるようになります。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB
本記事は以下の論文のフォローになります。
標的の選定方法からスクリーニング、スクリーニングした薬物候補化合物の毒性、物性予測、MD シミュレーションまでしており、ほぼ無料のツールですることができます。in silico創薬の基本が詰まっているので、一緒に勉強していきましょう。IFは3.8(2024年)の論文です。
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
In silico創薬とその流れ
In silico創薬、in silicoスクリーニングとは
インシリコ創薬(in silico drug discovery)は、コンピュータを駆使しし、シミュレーションやデータ解析を用いて新薬を設計・発見する手法です。
これにより、従来の実験的な方法に比べてコストと時間を大幅に削減できるとされています。
その中でもin silicoスクリーニングは、膨大なデータベースから有望な候補物質を迅速に特定し、その効果や副作用を予測することで、創薬の初期段階での効率化が図られます。
一見難しそうなin silico創薬ですが、現在では様々なアプリケーションやwebサイトがあり、それらを駆使すれば、誰でも簡単に創薬をすることもできます。本記事では、それらのアプリケーションを駆使し、in silico創薬を行った論文をもとに、手法をわかりやすく説明していきます。
In silico創薬の流れ
- ターゲット選定、準備:疾患の原因となる分子(ターゲット)を特定し、その3D構造を解析します。
- 化合物ライブラリの構築:in silicoスクリーニングに使用する化合物集団(=ライブラリ)を作成します。
- in silicoスクリーニング:コンピュータ上で数百万の化合物を対象に、ターゲット分子との結合親和性を評価します。
- ドッキングシミュレーション:候補化合物がターゲット分子にどのように結合するかを詳細にシミュレーションします。
- 分子動力学シミュレーション:候補化合物とターゲットの相互作用を動的に解析し、安定性や効果を予測します。
- 物性、毒性評価:過去のデータを用いて、新たな候補物質の特性や副作用を予測するモデルを構築します。
- (実験的検証):in silico解析で得られた候補物質を実験室で合成し、実際の効果や安全性を検証します。
本記事は1.のターゲット選定、準備を解説します。
これまでLabcodeで紹介してきた記事(PyRx, Autodock)に加え、準備したタンパク質の安定性のチェックの仕方も加えております。
以下の論文をフォローしていきます。
本記事で用いられるツール一覧
UCSF Chimera 1.17.3 :タンパク質可視化、準備に重要
PDBsum: タンパク質の安定性評価の目安であるRamachandran Plotの作成に使用
ターゲットの選定
今回ターゲットになる5-enolpyruvylshikimate-3-phosphate (EPSP) synthaseは過去の知見から選定されています。
Acinetobacter baumannii(A. baumannii)は多剤耐性を持つ「スーパー菌」として知られており、世界的に優先的に対策が求められている病原体です。この細菌は、免疫力が低下した人々の間で呼吸器感染、創傷感染、血流感染などを引き起こしやすいです。
A. baumanniiに対する治療法としては、コリスチンやタイゲサイクリンなどの薬剤が使用されていますが、耐性が増加しているため、より効果的な新しい薬物標的の探索が求められています。
この研究では、A. baumanniiの5-エノールピルビルシキミ酸-3-リン酸(EPSP)合成酵素がターゲットとして選ばれました。EPSP合成酵素はシキミ酸経路の一部であり、この経路は細菌に存在し、人間には存在しないため、細菌の成長と生存に必要不可欠な酵素です。このような特徴から、シキミ酸経路は新しい抗菌薬の標的として非常に魅力的です。
タンパク質の準備
論文のMaterials and methodsの「Retrieval EPSP synthase crystal structure and preparation for docking studies」の部分フォローしていきます。
まずは5BUFのPDBからPDBファイルをダウンロードしてください。
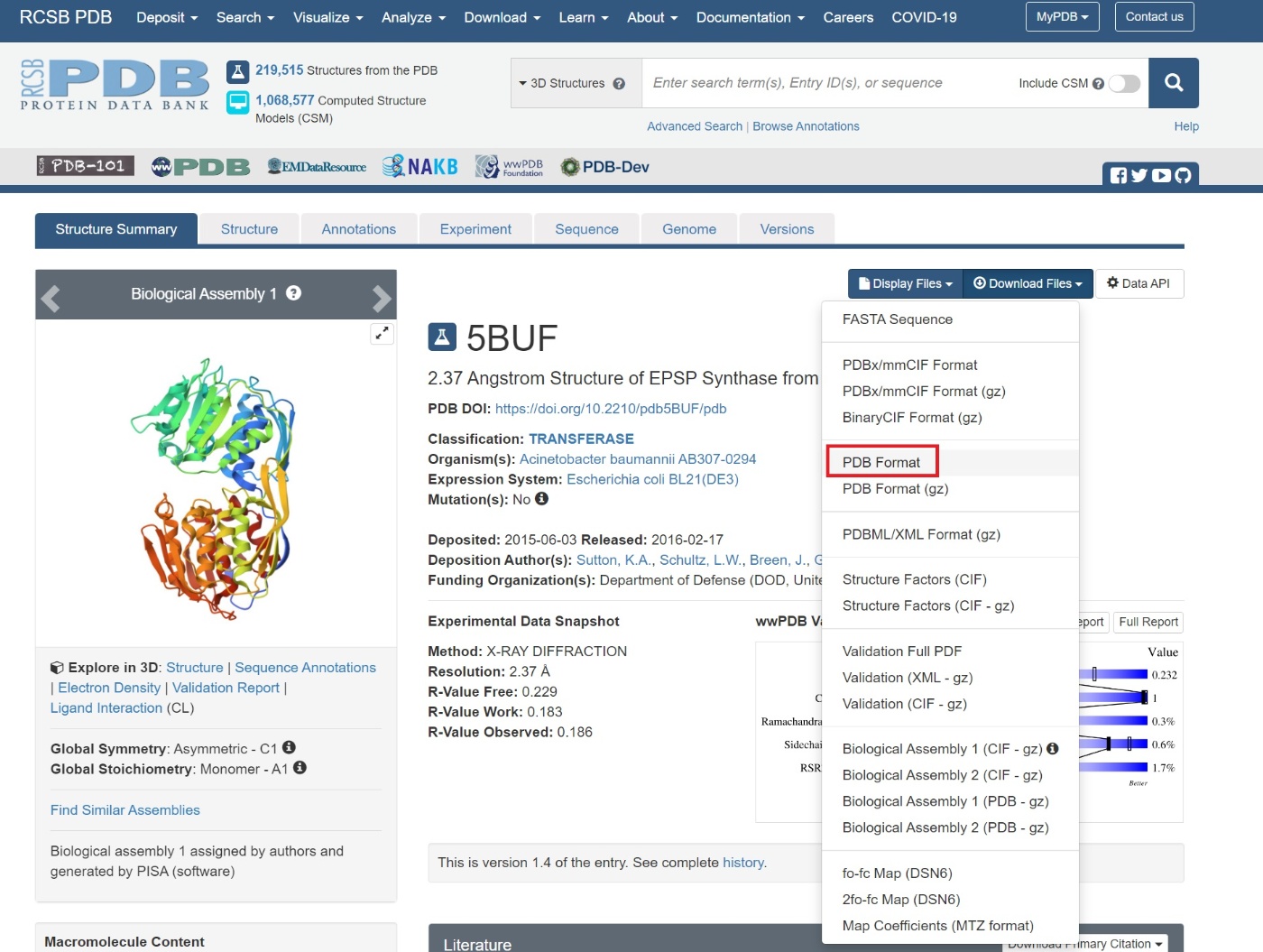
続いて、ダウンロードしたタンパク質の水分子を消していきます。(論文ではextra chainやリガンドも除くと書いてありますがありません)
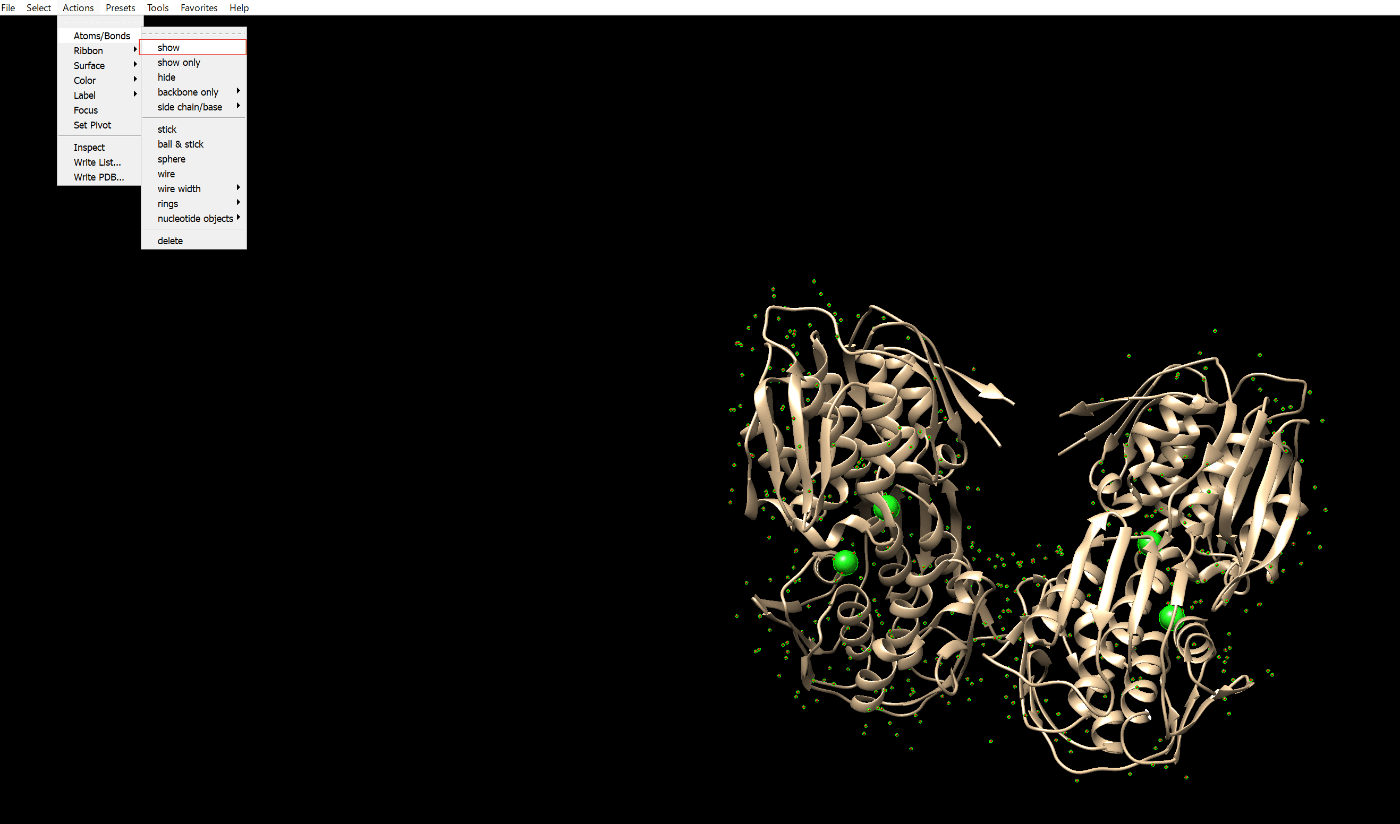
わかりやすいようにいったん水分子とイオンを表示しておきます。
UCSF ChimeraでダウンロードしたEPSPを開き、Actions→Atoms/Bonds→showですべての分子を表示します。
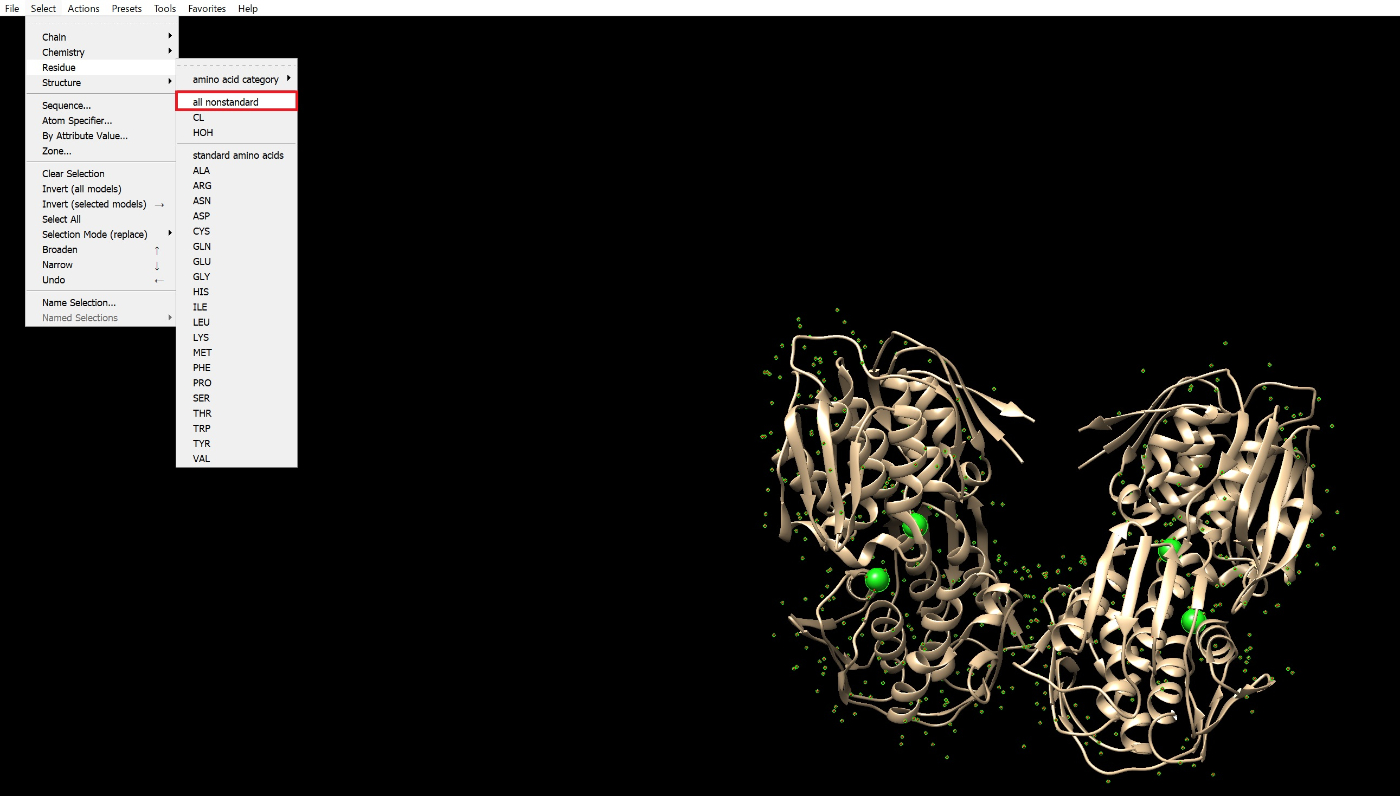
以下のようにSelect→Residue→all nonstandardを押し、アミノ酸以外の分子を選択してください。
続いて、Actions→Atoms/Bonds→deleteで選択した水分子、イオンを消します。
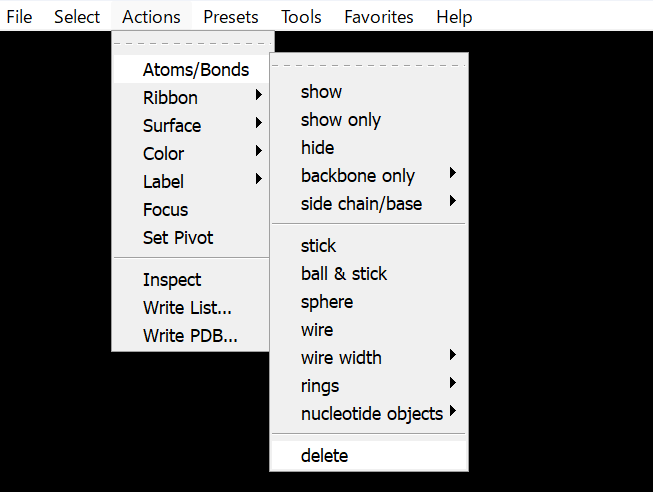
水分子とイオンが消えて、タンパク質のみになりました。
水素化、電荷付加
続いて、水素化、電荷の負荷を行います。
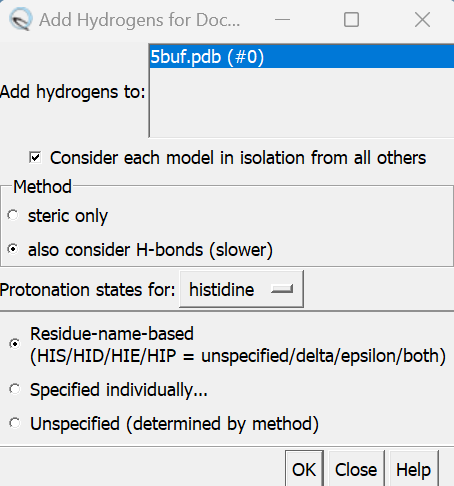
Tools→Structure Editing→Dock Prepを押してください。
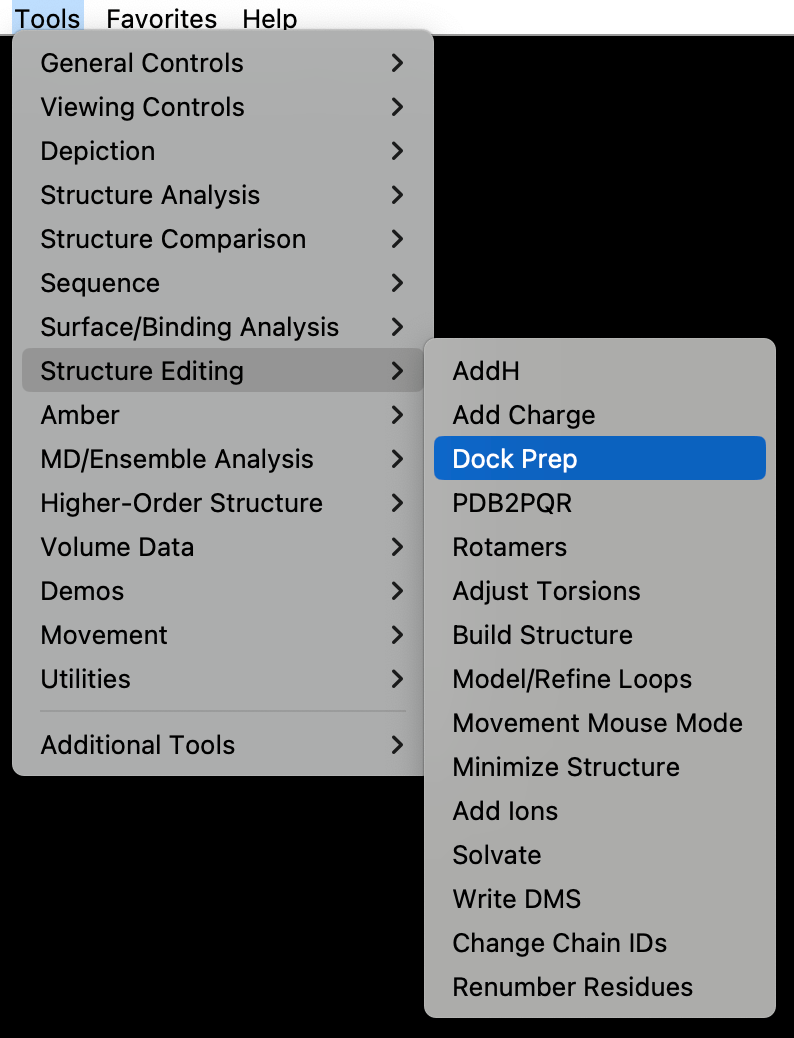
以下の画面が出ます。Add hydrogens, Add chargesにチェックがあることを確認して、OKを押してください。(他の箇所は今回はあまり影響ないので、そのままでいいです。)
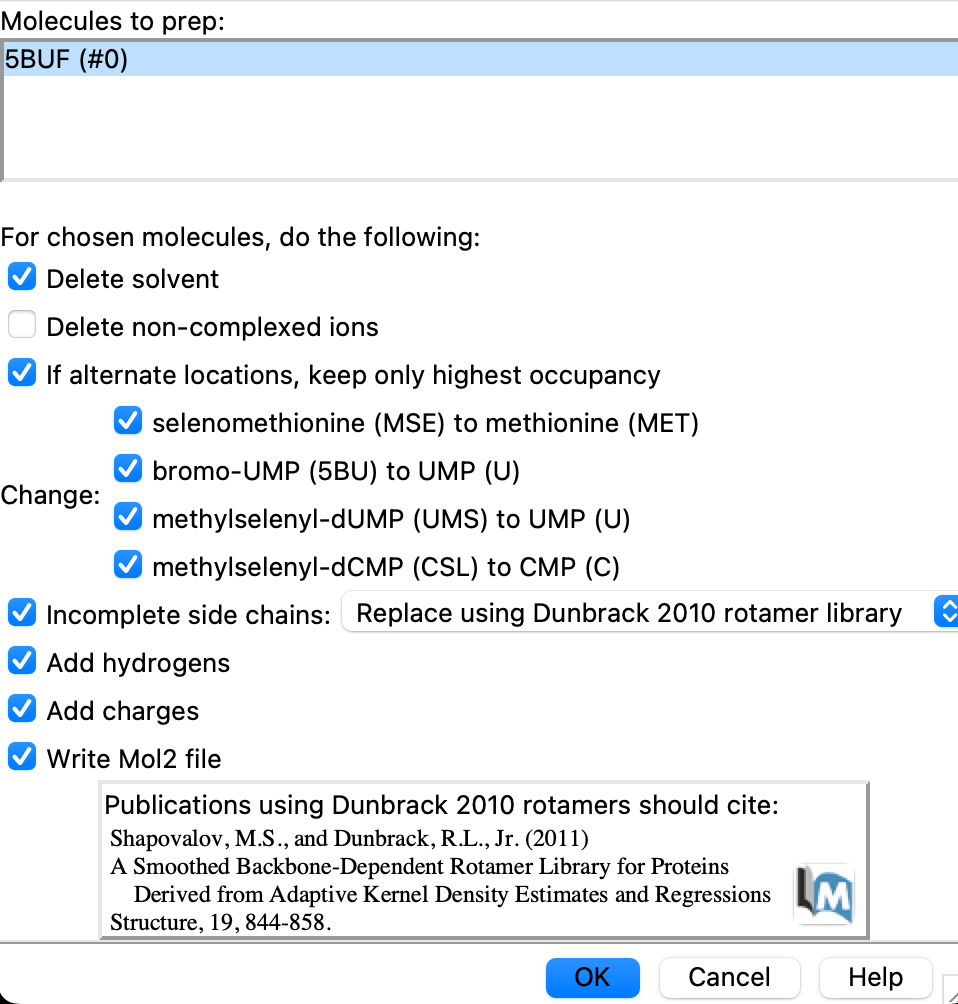
エネルギーの最小化
続いて、エネルギーの最小化を行います。PDBからダウンロードしたタンパク質の多くはリガンドや水分子、イオンが結合したものとなっており、上記の操作でリガンドを消した状態で最も安定な構造ではありません。なので、エネルギーの最小化の処理を行います。
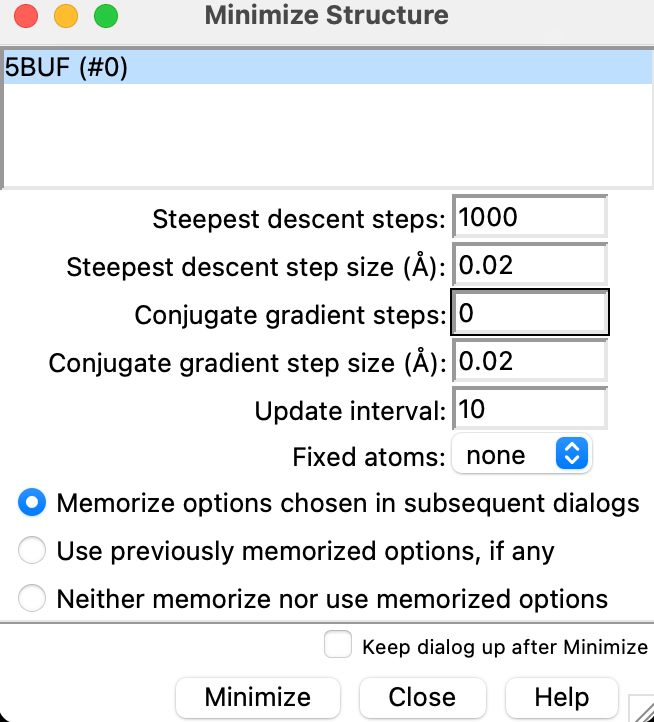
Tools→Structure Editing→Minimize Structureを押してください。
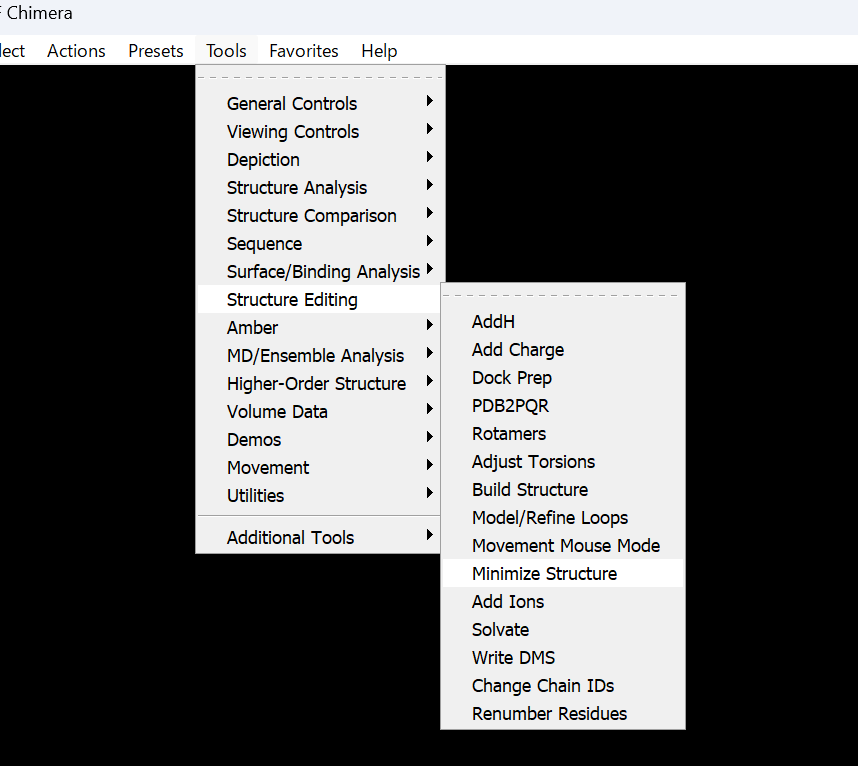
Steepest descent stepsを1000に設定してください。Conjugate gradient stepsも設定した方が良さそうですが、論文に合わすために、0にします。
設定したらMinimizeを押してください。
以下に説明を書きます。
-
エネルギー最小化:
- エネルギー最小化は、分子構造の不自然なひずみや異常を解消し、安定な構造を得るための計算手法です。このプロセスでは、1000回の最急降下法(steepest descent)と共役勾配法(conjugate gradient)のアルゴリズムを用います。
- 最急降下法は、最初に大きなエネルギー勾配を持つ箇所を迅速に調整する方法で、初期のエネルギー低減に有効です。一方、共役勾配法は、より細かいエネルギー最小化を行うために使用され、全体のエネルギーを安定させます。
- ステップサイズ(step size)は、エネルギー最小化の際に行う調整の大きさを示します。0.02Åという設定は、非常に小さな調整を行いながらエネルギーを低減させることを意味します。
-
AMBER ff14SB:
- AMBER ff14SBは、分子力学シミュレーションで使用される力場の一つであり、特にタンパク質のバックボーン(主鎖)とサイドチェーン(側鎖)の残基のパラメータ化に使用されます。これにより、分子シミュレーション中に適切な力が各原子に作用するように設定されます
続いて、以下の設定画面が出てくると思います。
こちらに関してはデフォルト設定で大丈夫なので、そのままOKを教えてください。
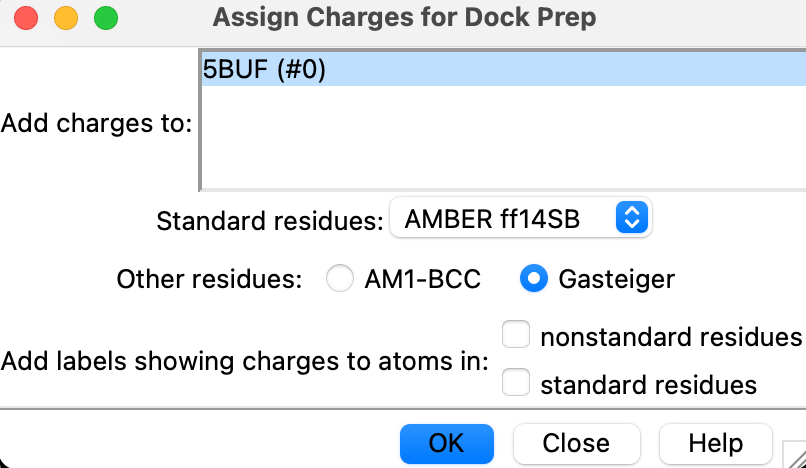
続いて、以下の画面が出ます。デフォルトの設定で大丈夫ですが、
Standard residuesをAMBER ff14SBであることを確認し、OKを押してください。
Other residuesについては今回は関係ないですが、Gasteigerにしてあることが多いので、それに設定しておきます。
左下に、エネルギー最小化の進捗がでてきます。1時間くらいかかります。
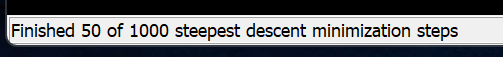
終わったら、PDBファイルを5BUF_minimization.pdbとして、保存してください。
Ramachandran plotの作成
最小化したタンパク質はRamachandoran plotで評価します。
Ramachandranプロットは、タンパク質の二次構造を評価するためのツールであり、各アミノ酸残基の二つの主鎖ジアヘドラル角度、φ (phi) と ψ (psi) をプロットします。これらの角度は、ポリペプチド鎖の折りたたみや構造形成に大きな影響を与えます。
Ramachandoran plotはPDB sumを用いて作成します。
まずはPDB sumに行き、最小化したタンパク質とemail adressを記入し、アップロードを押してください。1時間くらいで結果が返ってきます。
エネルギー最小化後のRamachandranプロットの確認では以下を注意してみます。
- 残基の多くが許容領域(特に、αヘリックスやβシートに対応する領域)に集中しているか
- 許容されない領域にある残基の数が少ないか
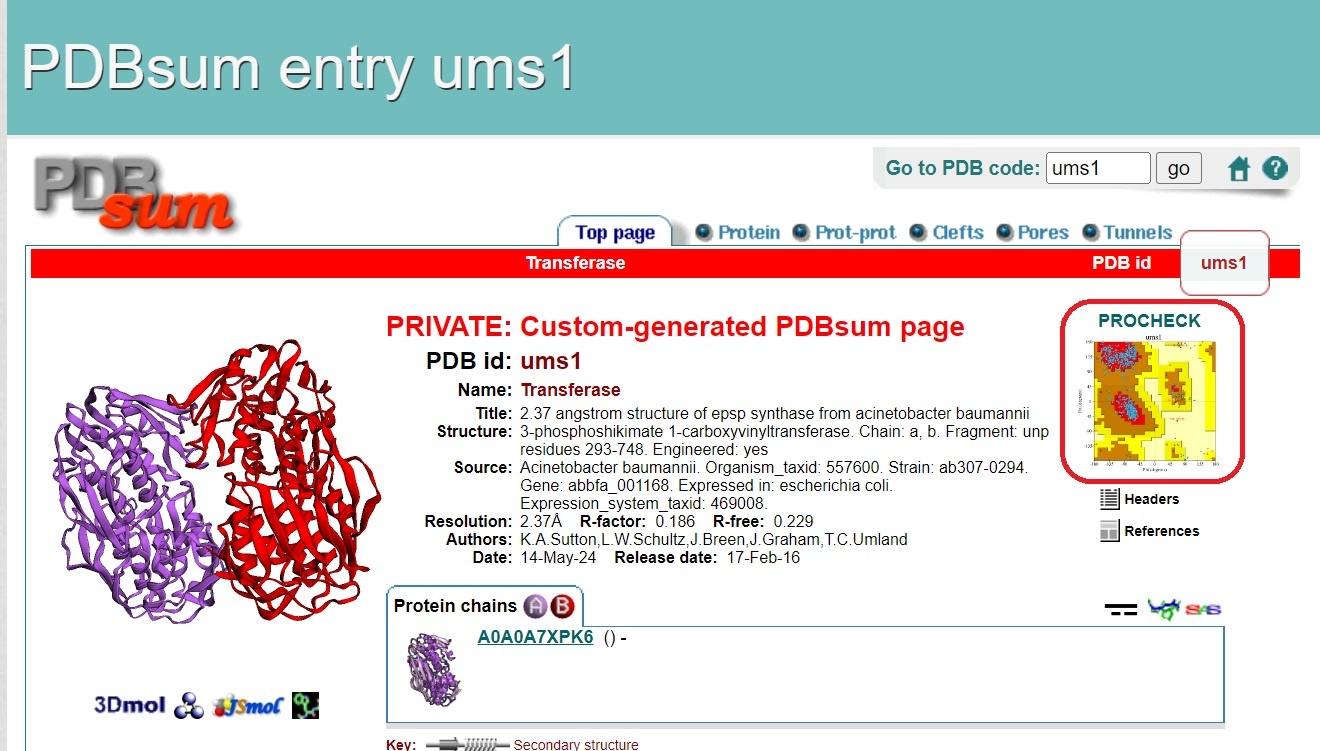
送られてきたメールを開くと以下のように表示されます。
右上にRamachandran Plotが表示されるので、開いてください。
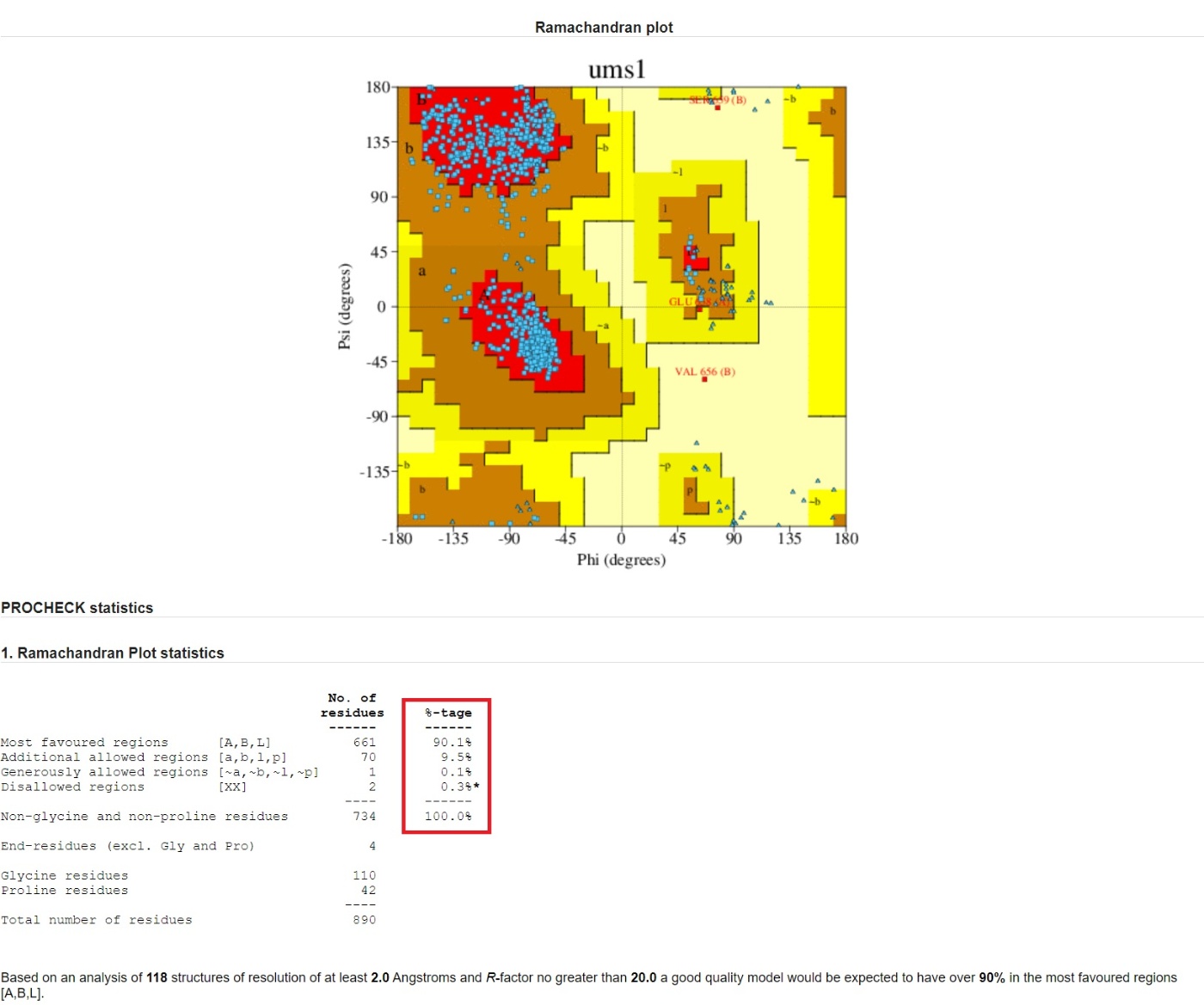
以下にRamachandran plotの見方について説明しします。
縦軸と横軸の説明
この画像は、Ramachandranプロットを示しています。これは、タンパク質の二次構造を評価するために使用される重要なツールです。
-
縦軸(Y軸):Psi (ψ) angle
縦軸は「Psi (ψ)」角を示しています。ψ角は、ポリペプチド鎖のCα原子に結合したカルボニル基と隣接するCα原子に結合したアミノ基の間の回転角度です。この角度は、−180度から180度までの範囲で示されます。
-
横軸(X軸):Phi (φ) angle
横軸は「Phi (φ)」角を示しています。φ角は、ポリペプチド鎖のCα原子に結合したアミノ基とその隣接するCα原子に結合したカルボニル基の間の回転角度です。この角度も、−180度から180度までの範囲で示されます。
見方の説明
Ramachandranプロットは、タンパク質の各アミノ酸残基のφ角とψ角の組み合わせを視覚的に表現するグラフです。具体的な見方を以下に説明します。
色分けされた領域:
- 赤色の領域: 最も好まれる領域を示しています。これらは、安定した二次構造(例えばαヘリックスやβシート)を形成するために必要なφ角とψ角の組み合わせを表しています。
- 茶色の領域: 追加の許容領域を示しており、これも比較的安定な構造を形成する角度の組み合わせです。
- 黄色の領域: 寛容される領域であり、多少許容されるが、あまり一般的ではないφ角とψ角の組み合わせです。
- 白色の領域: 禁じられた領域を示しています。これらの角度の組み合わせは、立体的な干渉が生じ、実際のタンパク質では非常にまれです。
赤枠内の情報
赤枠内には、各カテゴリに該当するアミノ酸残基の数("No. of residues")とその割合("%tage")が表示されています。これらのカテゴリは以下のように定義されています。
-
Most favoured regions [A,B,L]:
- No. of residues: 661
- %tage: 90.1%
- これは、Ramachandranプロット内の最も好ましい領域(赤色領域)に存在する残基の数と割合を示しています。この数値が高いほど、モデルが信頼できると考えられます。
-
Additional allowed regions [a,b,l,p]:
- No. of residues: 70
- %tage: 9.5%
- これは、追加で許容される領域(茶色の領域)に存在する残基の数と割合を示しています。これも比較的安定な構造を示しますが、最も好ましい領域よりは少し劣ります。
-
Generously allowed regions [~a,~b,~l,~p]:
- No. of residues: 1
- %tage: 0.1%
- これは、寛容される領域(黄色の領域)に存在する残基の数と割合を示しています。こちらの数値は低いほど良好なモデルといえます。
-
Disallowed regions [XX]:
- No. of residues: 2
- %tage: 0.3%
- これは、禁止された領域(白色の領域)に存在する残基の数と割合を示しています。この数値は低い方が望ましく、通常、信頼性の高い構造では禁止された領域に残基がほとんど含まれません。
PDBSumの結果、90.1%の残基が最も好ましい領域にあり、0.3%のみが禁止された領域にあります。このため、このタンパク質は非常に良好な品質であると考えられます。
以上でタンパク質の準備は終了です。
最後に
Youtubeなどではできるだけ簡単にするために、タンパク質準備はPDBからダウンロードしただけというものも多いと思います。
エネルギー最小化などをきちんと行い、できるだけ生体内に近いタンパク質の状態を保って、以後の分子ドッキングなどをやっていきましょう!
参考文献
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
Discussion