GROMACSによるMD simulation(分子動力学シミュレーション)のためのWindows環境の構築をやってみた
本記事はwslを使って、WindowsPCで分子動力学シミュレーションソフトのGROMACSを使えるようにするための環境構築方法を書いています。wslを使うとWindowsの中でLinuxが起動するので、GROMACSが使えるようになります。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB
本記事は以下の論文のフォローになりますが、論文には載っていないwindowsでのGROMACSの環境構築の部分です。
※本論文のMD simulationはAMBERを用いて行なっておりますが、本記事ではGROMACSでフォローしています。
Macの方は以前ご紹介したこちらの記事を参照していください。
本論文は標的の選定方法からスクリーニング、スクリーニングした薬物候補化合物の毒性、物性予測、MDシミュレーションまでしており、ほぼ無料のツールですることができます。in silico創薬の基本が詰まっているので、一緒に勉強していきましょう。
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
In silico創薬とその流れ
In silico 創薬、in silicoスクリーニングとは
インシリコ創薬(in silico drug discovery)は、コンピュータを駆使し、シミュレーションやデータ解析を用いて新薬を設計・発見する手法です。
これにより、従来の実験的な方法に比べてコストと時間を大幅に削減できるとされています。
その中でもin silicoスクリーニングは、膨大なデータベースから有望な候補物質を迅速に特定し、その効果や副作用を予測することで、創薬の初期段階での効率化が図られます。
一見難しそうなin silico創薬ですが、現在では様々なアプリケーションやwebサイトがあり、それらを駆使すれば、誰でも簡単に創薬をすることもできます。本記事では、それらのアプリケーションを駆使し、in silico創薬を行った論文をもとに、手法をわかりやすく説明していきます。
In silico創薬の流れ
- ターゲット選定、準備:疾患の原因となる分子(ターゲット)を特定し、その3D構造を解析します。
- 化合物ライブラリの構築:in silicoスクリーニングに使用する化合物集団(=ライブラリ)を作成します。
- in silicoスクリーニング:コンピュータ上で数百万の化合物を対象に、ターゲット分子との結合親和性を評価します。
- 分子動力学シミュレーション:候補化合物とターゲットの相互作用を動的に解析し、安定性や効果を予測します。
- 物性、毒性評価:過去のデータを用いて、新たな候補物質の特性や副作用を予測するモデルを構築します。
- (実験的検証):in silico解析で得られた候補物質を実験室で合成し、実際の効果や安全性を検証します。
すでに公開したものについては、リンクを貼っています。
本記事は4.の分子動力学シミュレーションのための環境構築で、Windowsの中にlinuxを入れていきます。
WindowsへのLinuxの導入
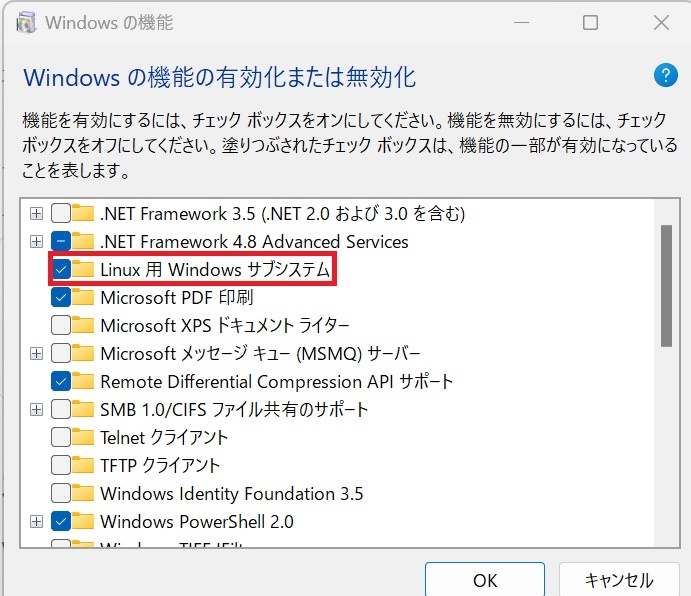
コントロールパネル→プログラム→Windowsの機能の有効化または無効化を開いてください。

以下のLinux用Windowsサブシステムにチェックが入っていることを確認してください。
続いて、Ubuntuのアプリをダウンロードしてください。
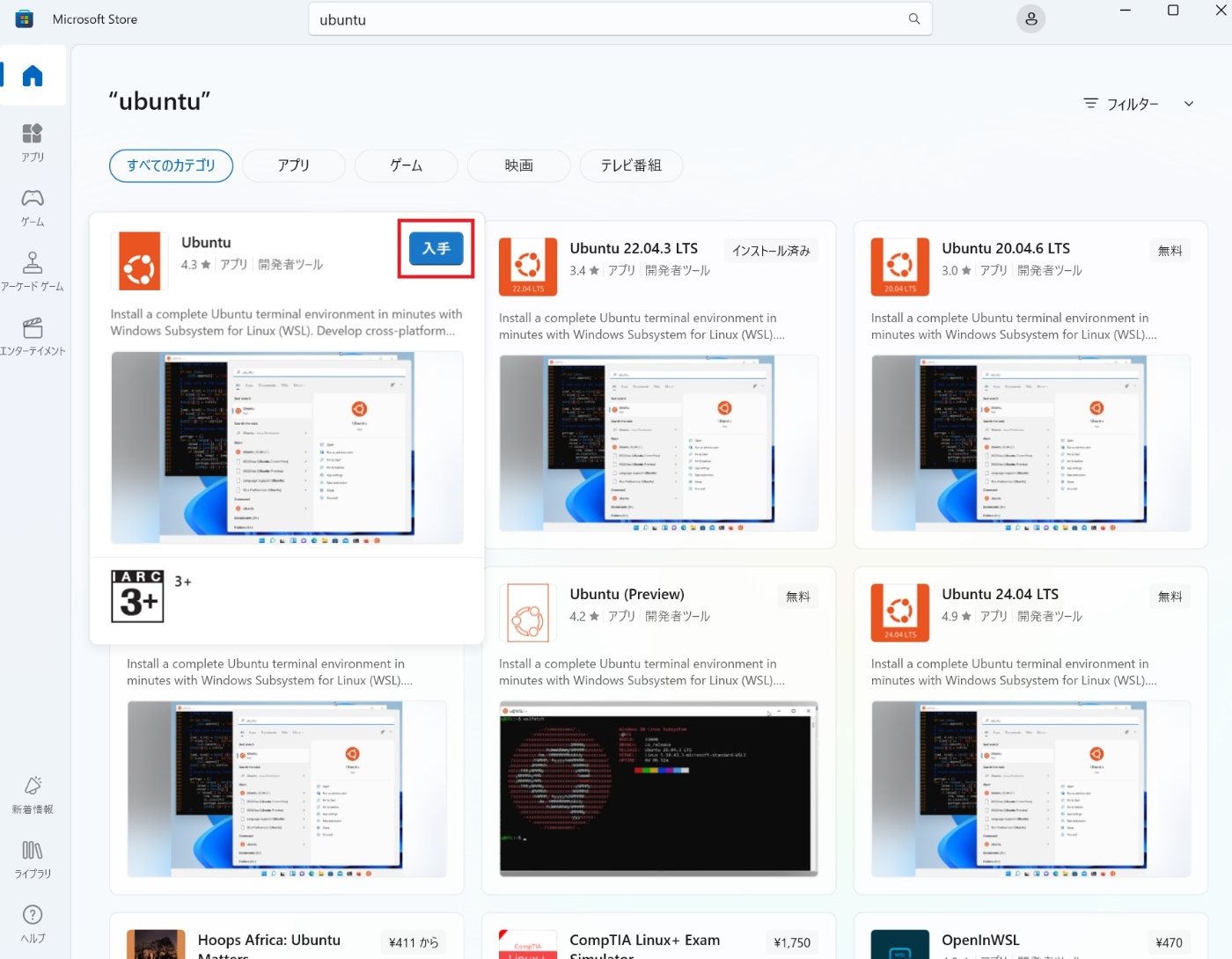
Ubuntuを開いて、以下のようにusernameとpasswordを設定してください。

これが成功すると、ubuntuが使えるようになります!
以下をubuntuのCUIでの実行してください。
そうすると、windowsシステムのデスクトップが作業ディレクトリになります。
cd /mnt/c/users/(あなたのユーザー名)/desktop

/mntは、LinuxやUnix系のオペレーティングシステムで、外部のファイルシステム(例えば、外付けハードドライブやUSBドライブなど)を一時的にマウントするための標準的なディレクトリです。/mntを使うことで、これらの外部デバイスにアクセスし、ファイルの読み書きができるようになります。
GROMACSのインストール
GROMACSのインストール方法を以下で説明します。
前半に作業手順だけを紹介し、後半で前半の作業について解説をします。
作業手順
まずcd を実行して、ホームディレクトリに戻ってください。
続いて、以下を順番に実行してください。
sudo apt-get update
sudo apt-get upgrade
sudo apt-get install cmake
sudo apt install build-essential
mkdir gromacs/
cd gromacs/
wget https://ftp.gromacs.org/regressiontests/regressiontests-2024.tar.gz
tar xvzf regressiontests-2024.tar.gz
sudo apt-get install libfftw3-dev
mkdir build
cd build
(CPUのみを使う方)
sudo cmake .. -DGMX_BUILD_OWN_FFTW=OFF -DREGRESSIONTEST_DOWNLOAD=OFF -DCMAKE_C_COMPILER=gcc -DREGRESSIONTEST_PATH=./../../regressiontests-2024
make check
sudo make install
(GPUを使う方)
sudo apt install nvidia-cuda-toolkit
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DGMX_GPU=CUDA -DCMAKE_INSTALL_PREFIX=/usr/local/gromacs -DGMX_MPI=OFF -DCUDA_TOOLKIT_ROOT_DIR=/usr/lib/x86_64-linux-gnu -DCUDA_ARCHITECTURES=86
make -j 8
sudo make install
source /usr/local/gromacs/bin/GMXRC
echo "source /usr/local/gromacs/bin/GMXRC" >> ~/.bashrc
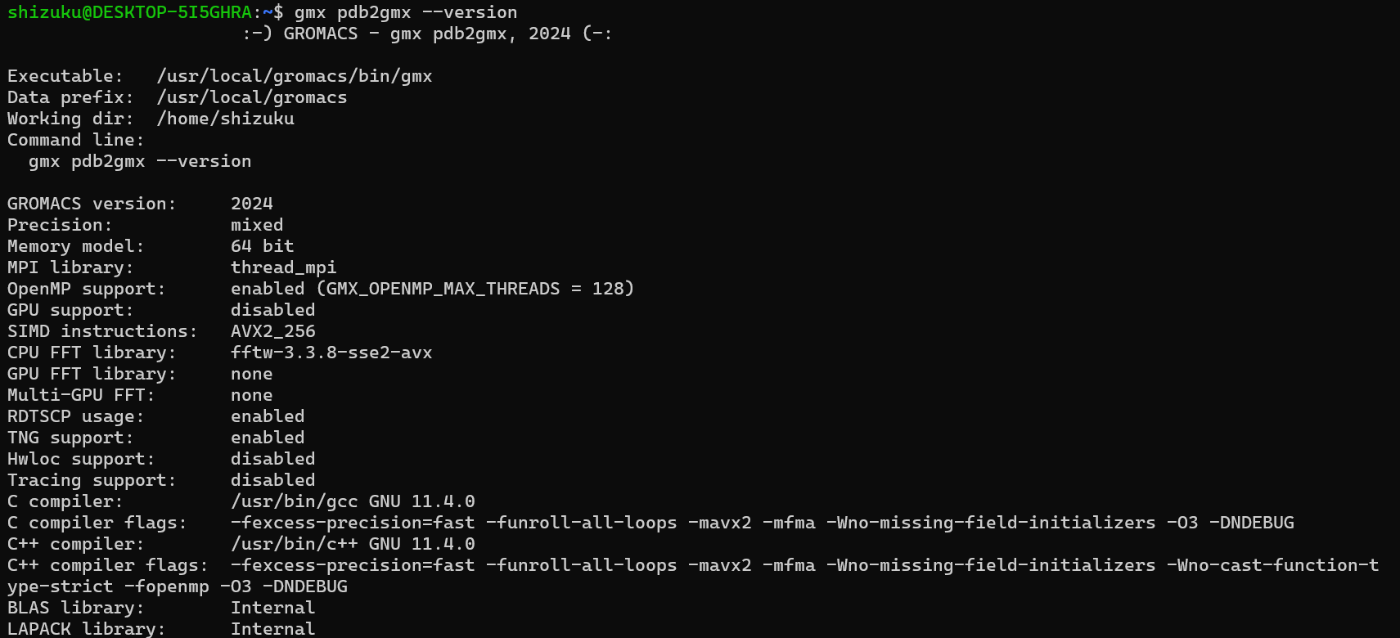
gmx pdb2gmx --version
以下のように出力されれば、OKです!
解説
sudo apt-get update
このコマンドは、パッケージインデックスを更新します。具体的には、リポジトリから最新のパッケージリストを取得し、ローカルに保存されているリストを更新します。
-
sudo:管理者権限でコマンドを実行します。 -
apt-get:パッケージ管理ツールです。 -
update:パッケージリストを更新するサブコマンドです。
実行後、システムは最新のパッケージ情報を持つことになり、新しいソフトウェアや更新されたソフトウェアの情報を知ることができます。
sudo apt-get upgrade
このコマンドは、updateで取得した最新のパッケージリストに基づいて、システムにインストールされているすべてのパッケージをアップグレードします。
-
sudo:管理者権限でコマンドを実行します。 -
apt-get:パッケージ管理ツールです。 -
upgrade:インストールされているすべてのパッケージを最新バージョンにアップグレードするサブコマンドです。
実行後、システムは可能な限り最新のパッケージで構成されるようになります。ただし、パッケージの依存関係が変更された場合など、いくつかのパッケージはアップグレードされないことがあります。
sudo apt-get install cmake
cmakeをインストールします。
ビルドプロセスとは、プログラムのソースコードを実行可能な形式(バイナリ)に変換する一連の手順のことです。CMakeは、GROMACSをビルドするために必要な設定、依存関係の管理、マルチプラットフォームのサポートを提供し、ビルドプロセスを簡単にするツールです。
sudo apt install build-essential
このコードはLinuxでプログラムを作成したり、ソースコードからソフトウェアをインストールしたりするために必要なツールをまとめてインストールします。
mkdir gromacs/
cd gromacs/
mkdir gromacs/でgromacsのディレクトリを作り、 cd gromacs/でそのディレクトリに入ります。
wget https://ftp.gromacs.org/regressiontests/regressiontests-2024.tar.gz
tar xvzf regressiontests-2024.tar.gz
wget https://ftp.gromacs.org/regressiontests/regressiontests-2024.tar.gz**:
-
wgetはインターネットからファイルをダウンロードするコマンドです。 - このコマンドでは、指定されたURLから
regressiontests-2024.tar.gzという名前のファイルをダウンロードします。このファイルは、GROMACSのテストファイルです。
tar xvzf regressiontests-2024.tar.gz:
-
tarはアーカイブファイル(複数のファイルをまとめたもの)を操作するコマンドです。 -
xvzfというオプションは、それぞれ以下の意味があります:-
x: ファイルを展開する(解凍)。 -
v: 展開中のファイル名を表示する(verbose)。 -
z: gzipで圧縮されたファイルを解凍する。 -
f: 処理するファイルを指定する。
-
- このコマンドでは、ダウンロードした
regressiontests-2024.tar.gzファイルを解凍して、その中身を取り出します。
sudo apt-get install libfftw3-dev
このコードは、FFTW(Fastest Fourier Transform in the West)ライブラリの開発ヘッダーファイルおよびライブラリをインストールするためのコマンドです。FFTWは、高速フーリエ変換(FFT)を実行するためのライブラリで、科学計算やシミュレーションに広く使用されています。
wget https://ftp.gromacs.org/gromacs/gromacs-2024.tar.gz
tar xvzf gromacs-2024.tar.gz
cd gromacs-2024/
このコマンドを順番に実行することで、GROMACSのソースコードをダウンロードし、そのソースコードのディレクトリに移動することができます。その後、GROMACSをビルドおよびインストールするための手順に進むことができます。
mkdir build
cd build
sudo cmake .. -DGMX_BUILD_OWN_FFTW=OFF -DREGRESSIONTEST_DOWNLOAD=OFF -DCMAKE_C_COMPILER=gcc -DREGRESSIONTEST_PATH=./../../regressiontests-2024
make check
sudo make instal
mkdir build, cd build でbuildディレクトリを作成し、buildディレクトリの中に入ります。
sudo cmake .. -DGMX_BUILD_OWN_FFTW=OFF -DREGRESSIONTEST_DOWNLOAD=OFF -DCMAKE_C_COMPILER=gcc -DREGRESSIONTEST_PATH=./../../regressiontests-2024
-
sudo cmake ..:cmakeはソフトウェアのビルド(コンパイル)設定を行うツールです。..はプロジェクトのルートディレクトリを指定します。sudoを使っているので、管理者権限で実行されます。 -
DGMX_BUILD_OWN_FFTW=OFF: GROMACS用のFFTWライブラリ(高速フーリエ変換のためのライブラリ)を自分でビルドせず、システムにあるものを使う設定です。 -
DREGRESSIONTEST_DOWNLOAD=OFF: GROMACSの回帰テストをダウンロードしない設定です。 -
DCMAKE_C_COMPILER=gcc: C言語のコンパイラとしてgccを使用することを指定しています。 -
DREGRESSIONTEST_PATH=./../../regressiontests-2024: 先ほどダウンロードしたregressiontests-2024のパスを指定しています。これにより、テストが実行できるようになります。
make check
make は、ソフトウェアをコンパイルするためのコマンドです。check は、ビルドしたソフトウェアが正しく動作するかをテストするためのターゲットです。このコマンドにより、GROMACSが正しく構築されているかどうかを確認します。
sudo make install
make でビルドしたソフトウェアをシステムにインストールします。sudo があるので、管理者権限でインストールが実行されます。このコマンドにより、GROMACSがシステムの適切なディレクトリにインストールされ、他の場所からも使えるようになります。
GPUを使う方はcmakeを以下に変更してください。
※GPUは使うと、MD simulationは1/10くらいになります。搭載している人は使いましょう!
sudo apt install nvidia-cuda-toolkit cmake .. -DGMX_BUILD_OWN_FFTW=ON -DGMX_GPU=CUDA -DCMAKE_INSTALL_PREFIX=/usr/local/gromacs -DGMX_MPI=OFF -DCUDA_TOOLKIT_ROOT_DIR=/usr/lib/x86_64-linux-gnu -DCUDA_ARCHITECTURES=86 make -j 8 sudo make install
source /usr/local/gromacs/bin/GMXRC
echo "source /usr/local/gromacs/bin/GMXRC" >> ~/.bashrc
gmx pdb2gmx --version
source /usr/local/gromacs/bin/GMXRC
-
source: シェル(コマンドライン)で、指定されたスクリプトを現在のシェル環境で実行するためのコマンドです。 -
/usr/local/gromacs/bin/GMXRC: GROMACSの環境設定スクリプトです。これを実行すると、GROMACSを使うために必要な環境変数(例えば、PATHなど)が設定されます。これにより、ターミナルでgmxコマンドを使えるようになります。
echo "source /usr/local/gromacs/bin/GMXRC" >> ~/.bashrc
-
echo "source /usr/local/gromacs/bin/GMXRC": 先ほどのsourceコマンドを実行するための行を作成します。 -
>> ~/.bashrc:~/.bashrcというファイルにその行を追加します。~/.bashrcは、ユーザーのシェルが起動されるたびに実行されるスクリプトファイルです。これにより、今後ターミナルを開くたびに自動的にGROMACSの環境が設定されます。
gmx pdb2gmx --version
-
gmx: GROMACSを操作するためのコマンドです。 -
pdb2gmx: GROMACSに含まれるツールの一つで、PDB形式のファイルからGROMACSで使える形式に変換するためのものです。 -
-version:pdb2gmxコマンドのバージョン情報を表示します。このコマンドを実行することで、GROMACSが正しくインストールされ、環境設定もできているかを確認できます。
以上でgromacsを使えるようにするためのwindows環境の構築は終わりです!
参考文献
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
Discussion