インシリコ創薬の大規模In silico screeningを論文に沿ってやってみる。【in silico創薬】
本記事は約7000個の低分子化合物を標的タンパク質に分子ドッキングすることで、低分子化合物のスクリーニングを行った記事です。
本記事の内容を試すことで、分子ドッキングという手法を使って大規模ライブラリから結合親和性が高い化合物を選び出せるようになります。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB
本論文は以下の論文のフォローになります。
標的の選定方法からスクリーニング、スクリーニングした薬物候補化合物の毒性、物性予測、MDシミュレーションまでしており、ほぼ無料のツールですることができます。in silico創薬の基本が詰まっているので、一緒に勉強していきましょう。
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
In silico創薬とその流れ
In silico 創薬、in silicoスクリーニングとは
インシリコ創薬(in silico drug discovery)は、コンピュータを駆使し、シミュレーションやデータ解析を用いて新薬を設計・発見する手法です。
これにより、従来の実験的な方法に比べてコストと時間を大幅に削減できるとされています。
その中でもin silicoスクリーニングは、膨大なデータベースから有望な候補物質を迅速に特定し、その効果や副作用を予測することで、創薬の初期段階での効率化が図られます。
一見難しそうなin silico創薬ですが、現在では様々なアプリケーションやwebサイトがあり、それらを駆使すれば、誰でも簡単に創薬をすることもできます。本記事では、それらのアプリケーションを駆使し、in silico創薬を行った論文をもとに、手法をわかりやすく説明していきます。
In silico創薬の流れ
- ターゲット選定、準備:疾患の原因となる分子(ターゲット)を特定し、その3D構造を解析します。
- 化合物ライブラリの構築:in silicoスクリーニングに使用する化合物集団(=ライブラリ)を作成します。
- in silicoスクリーニング:コンピュータ上で数百万の化合物を対象に、ターゲット分子との結合親和性を評価します。
- ドッキングシミュレーション:候補化合物がターゲット分子にどのように結合するかを詳細にシミュレーションします。
- 分子動力学シミュレーション:候補化合物とターゲットの相互作用を動的に解析し、安定性や効果を予測します。
- 物性、毒性評価:過去のデータを用いて、新たな候補物質の特性や副作用を予測するモデルを構築します。
- (実験的検証):in silico解析で得られた候補物質を実験室で合成し、実際の効果や安全性を検証します。
すでに公開したものについては、リンクを貼っています。
本記事は3. in silicoスクリーニングを解説します。
低分子化合物のエネルギーの最小化とSite directed virtual screening(SDVS)をやっていきます。
スクリーニングシステムの一つGOLDは有料のソフトウェアなので、今回はPyRXのみによるスクリーニングになります。
本記事で用いられるツール一覧
PyRx: In silicoスクリーニング用
Discovery Studio: 結果の可視化用
PyRxによる化合物ライブライブラリのエネルギー最小化
PyRxのinstall
まずPyRXのダウンロードページからwindows版のFree 0.8をダウンロードします。

ダウンロードしたPyRx-0.8-Setup.exeからPyRxをインストールしてください。特に特別なことはせず、Nextを押してインストールしてください。
PyRx内での低分子のエネルギー最小化
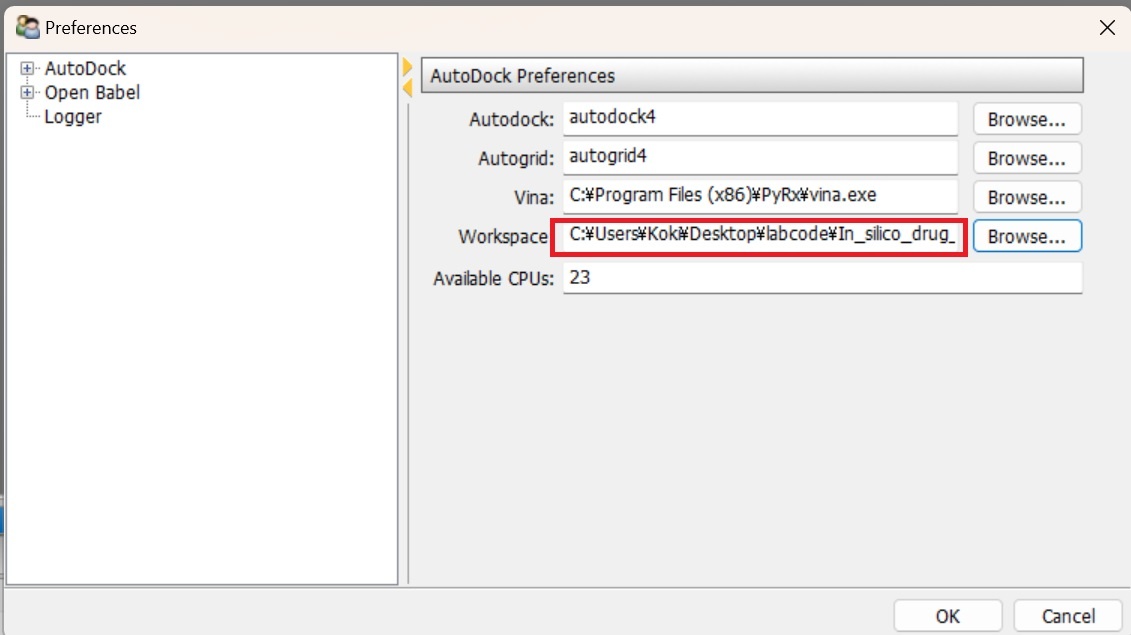
まずは忘れないうちにoutputディレクトリを決めておきます。
PyRxを開き、Edit→Preferencesを押してください。
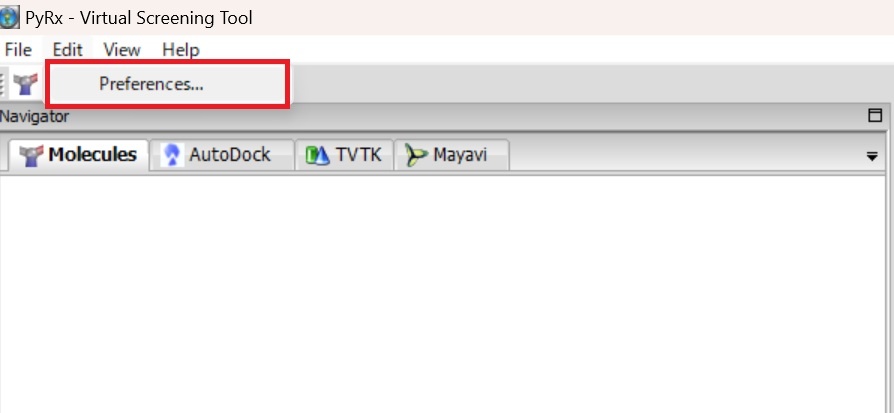
以下のworkspaceに任意のディレクトリを設定してください。これがOutputディレクトリとなります。
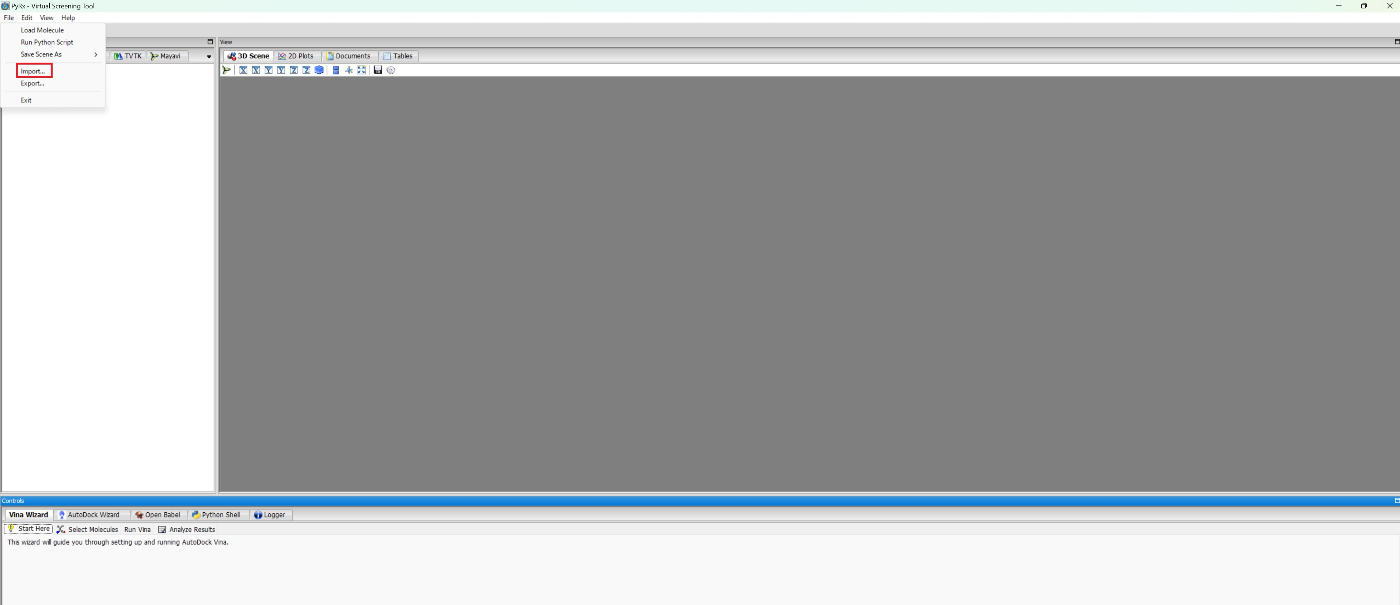
続いて、フィルタリングしたライブラリーを取り込んでいきます。
File→Importを押してください。
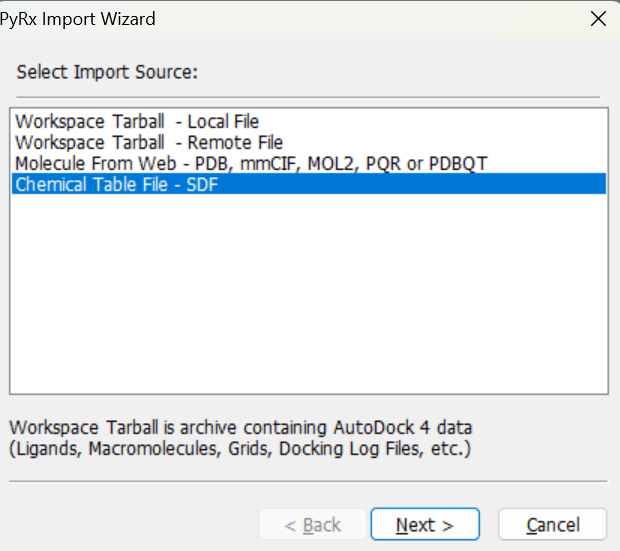
SDFを押して、フィルタリングしたCMNPD-filtered をアップロードしてください。
以下のようにアップロードできたと思うので、すべての化合物が入っているか確認してください。
Open Babelのタブのところにあります。
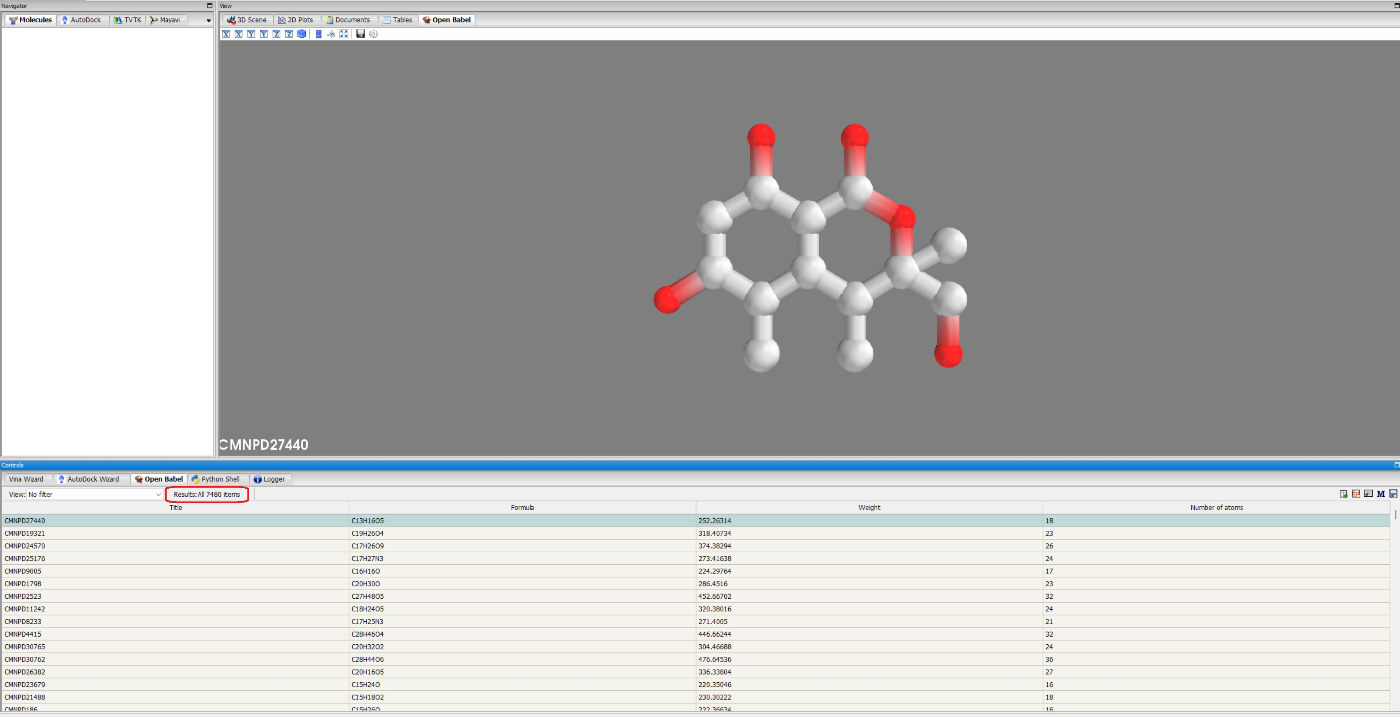
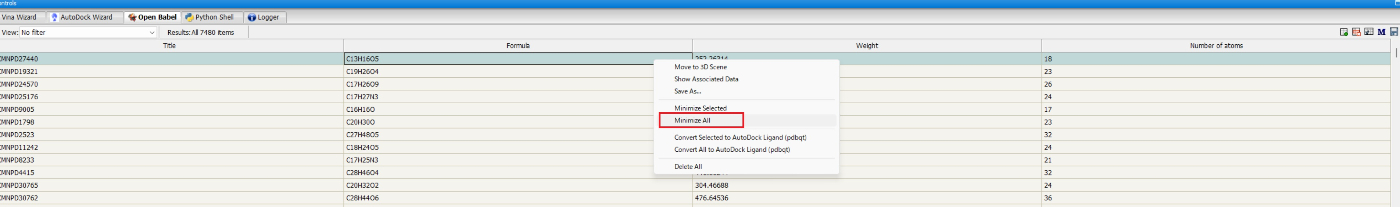
適当な化合物に対して、右クリックを押し、すべての低分子化合物のエネルギーを最小化します。
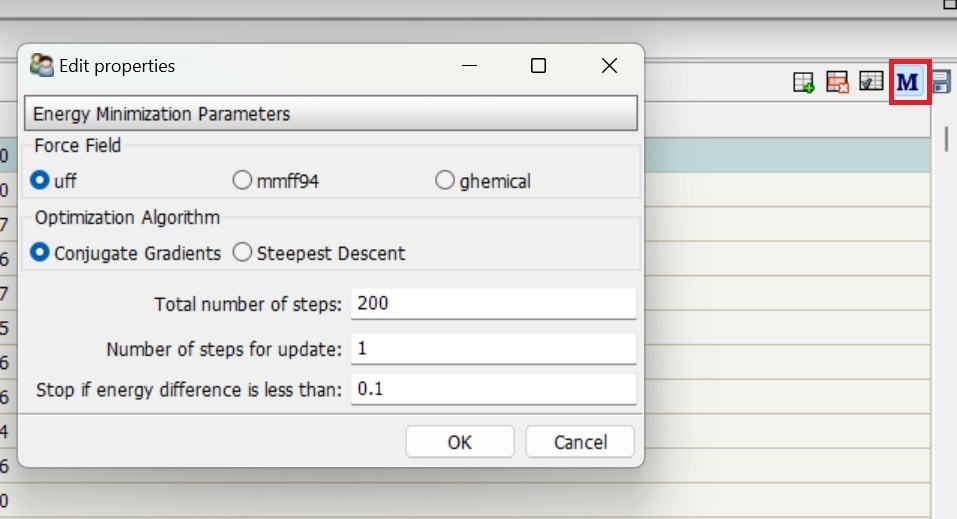
論文にはエネルギー最小化の方法が書かれていないので、デフォルトのuffで行います。
Force Fieldの変更の仕方
他のFocve Fieldで行いたい方は、以下の画像の右上にあるMを押して、一つを別の方法でエネルギー最小化を行った後、再度Minimize Allを行ってください。
Estimated timeは15分くらいですが、だいたい30分くらいかかります。

続いて、すべてのファイルをpdbqtファイルに変換します。
Estimated timeは15分くらいですが、だいたい1時間くらいかかります。
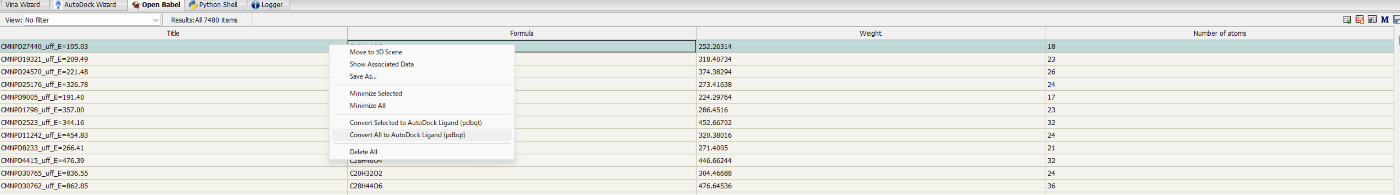
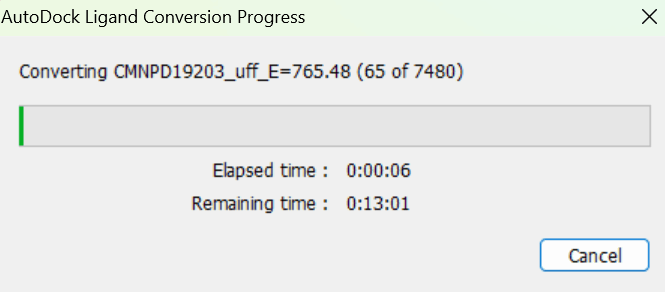
これで化合物ライブラリの準備は終了しました!
In silicoスクリーニング
用意したタンパク質とエネルギー最小化した化合物ライブラリを設定します。
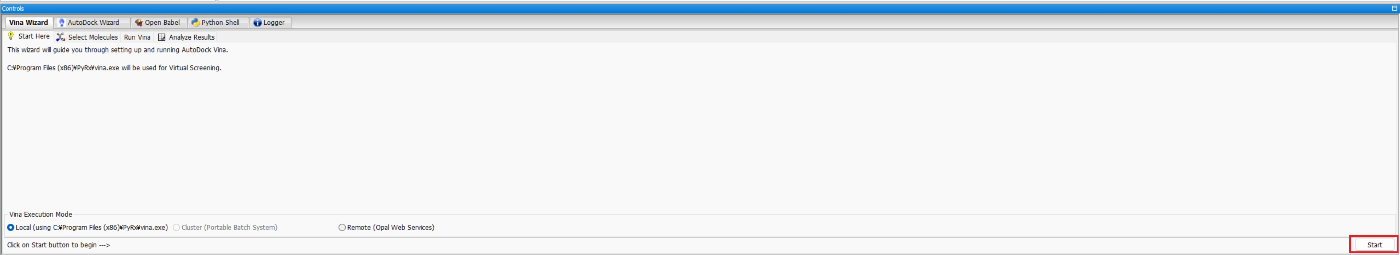
Vina WizardタブでStartを教えてください
Add Macromoleculeからエネルギー最小化したタンパク質をアップロードします。
アップロードするとタンパク質が表示されます。

アップロードしたタンパク質をクリックします。
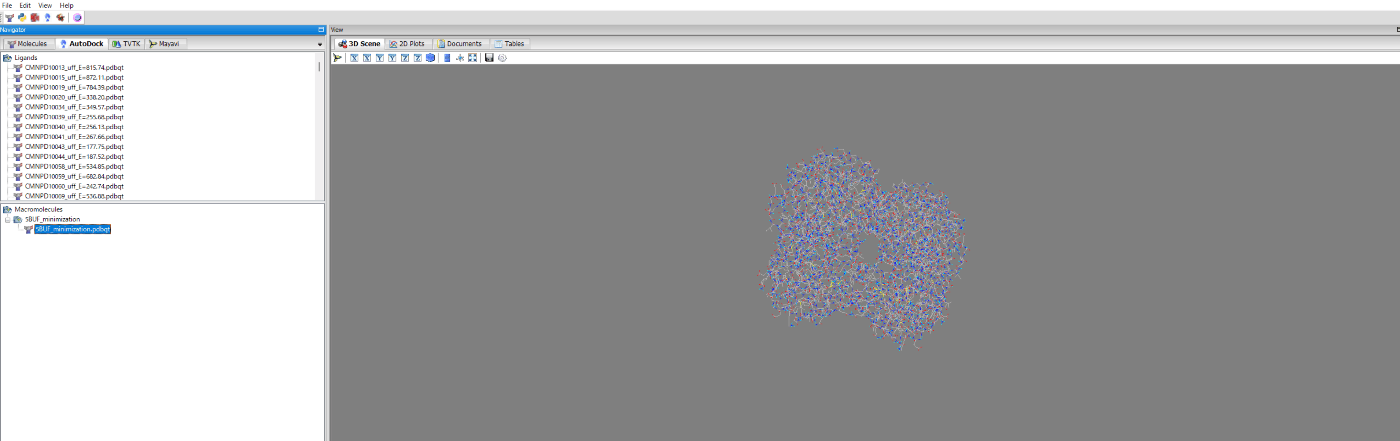
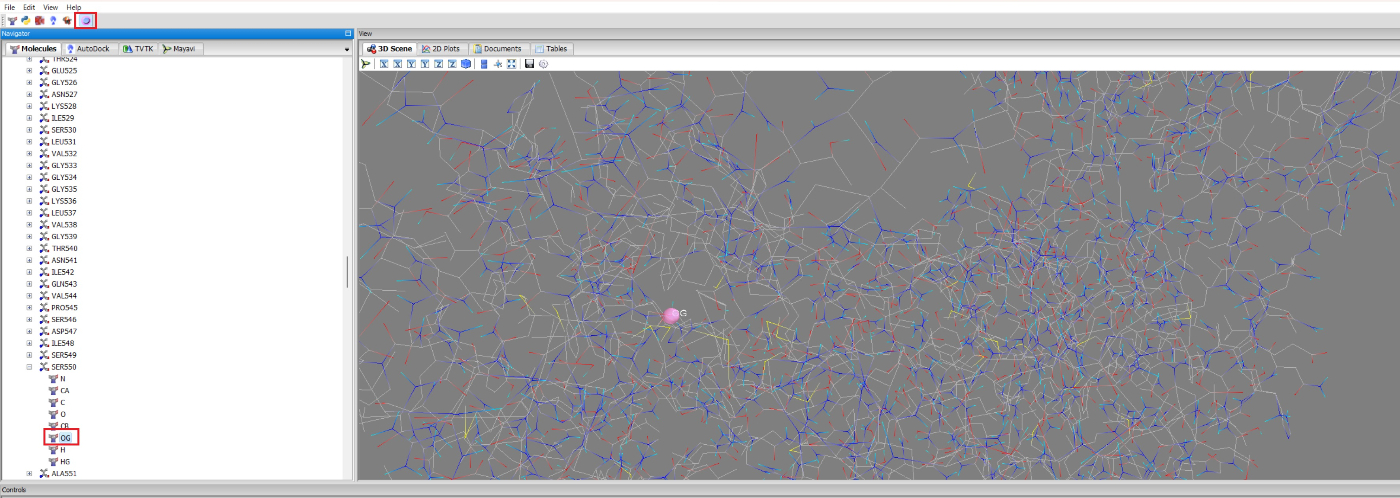
そうすると、Moleculeタブで各アミノ酸が設定されるので、Ser550 OG (論文ではSer238 OG、アミノ酸開始が1なので。)をクリックして、マークしてください。後で、ドッキング領域を決めるGridboxの目安となります。
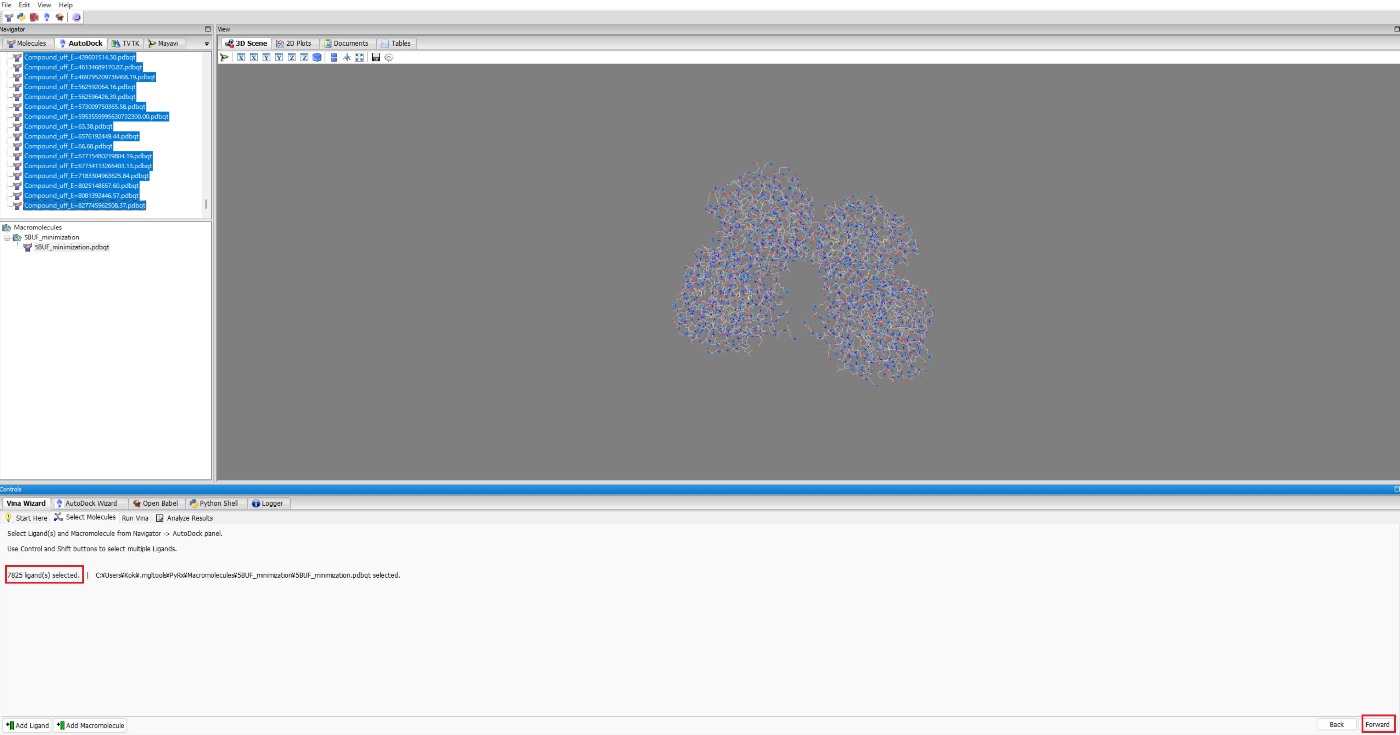
その後、すべてリガンドを選択して、Forwardを押してください。
リガンドを選択したとき、リガンドの数が少し減っているかと思います。
これはpdbqtに変換したときにエラーしたものが、含まれていないためです。
スクリーニングの段階なので、とりあえずは無視して先に進みます。
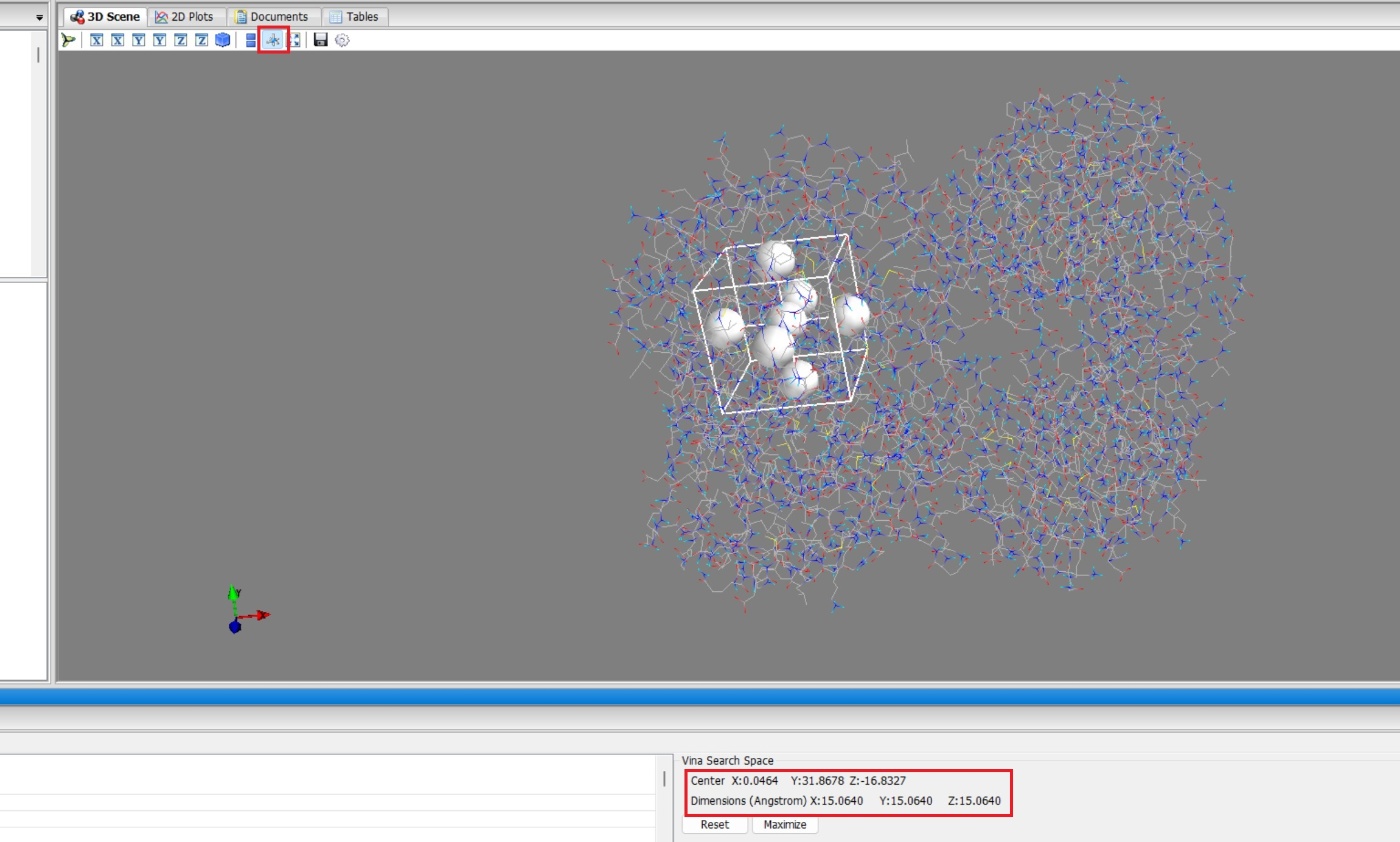
続いて、Grid Boxを決めていきます。
だいたい論文で定義してある値に揃えてください。
先ほど印をつけたSer550のOGアトムが真ん中に来るようにGrid Boxを設定してください。
X-axis: −0.544 Å, Y-axis: 28.321 Å, and Z-axis: −12.451 Åになるべく合うようにCenterを微調整してください。
以下の図の左上に座標のアイコンがあるので、微調整はそれを参考にすると少しわかりやすいです。Angstromのx、y、zは15くらいに設定してください。
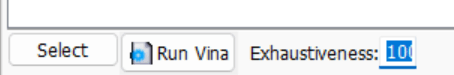
論文には100 iterationと書いてありますが、他の論文を読むと左下にあるEffectivenessの値っぽいので、この値を100に合わせておきます。(直訳の通り、100回繰り返したのかもしれませんが、その場合もこちらでは省略します)
右下のForwardをもしくは下のRunVinaを押すと分子ドッキングが始まります。

実行画面は以下のような感じです。
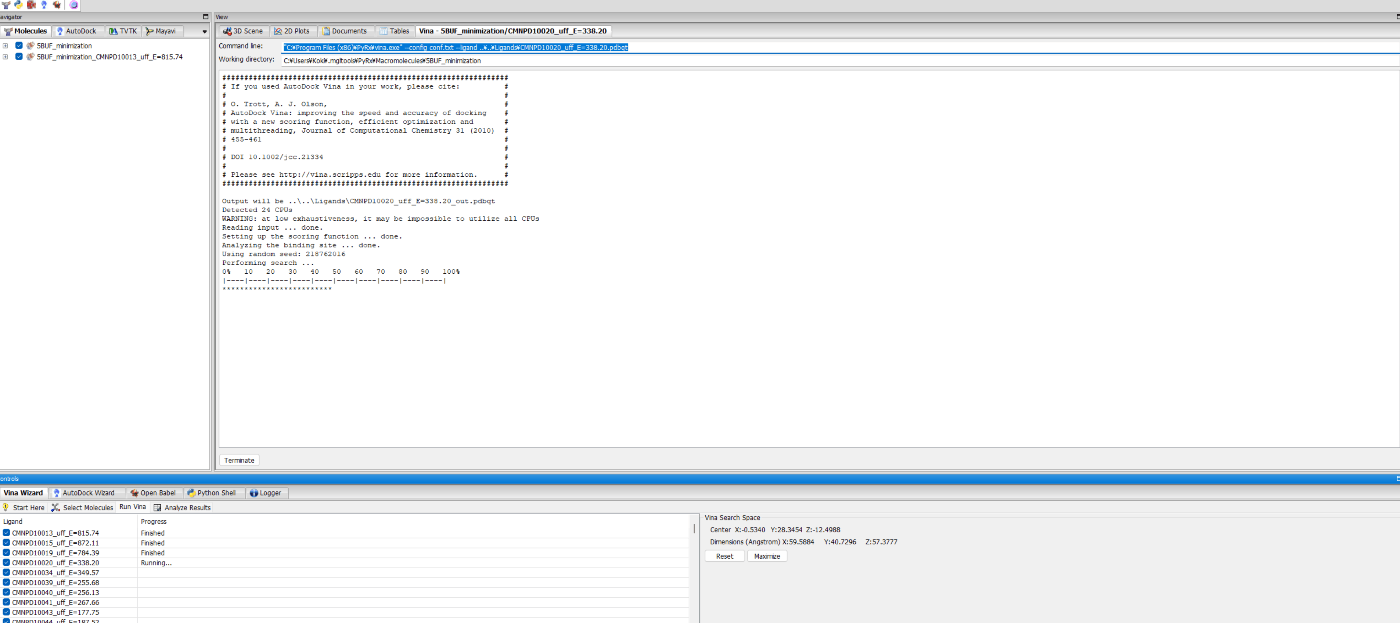
ちなみにスクリーニング中の使用メモリは1GBくらいです。意外と少ないです。

一晩くらいかかるので、気軽に待ちましょう。
6000くらいでPyRxが落ちてしまうため、半分くらいで二回に分けるとよいっでしょう。
結果
PyRxは結構落ちやすいソフトなので、結果が出たら、早めにsaveしてください。結果は最初に設定したOutputフォルダにあるので、途中で落ちた場合は途中のリガンドから再開すると良いでしょう。
結果の右上のsaveボタンでsaveできます。
Binding Energyの所を箇所を押して、エネルギーが低い順に並び替えます。
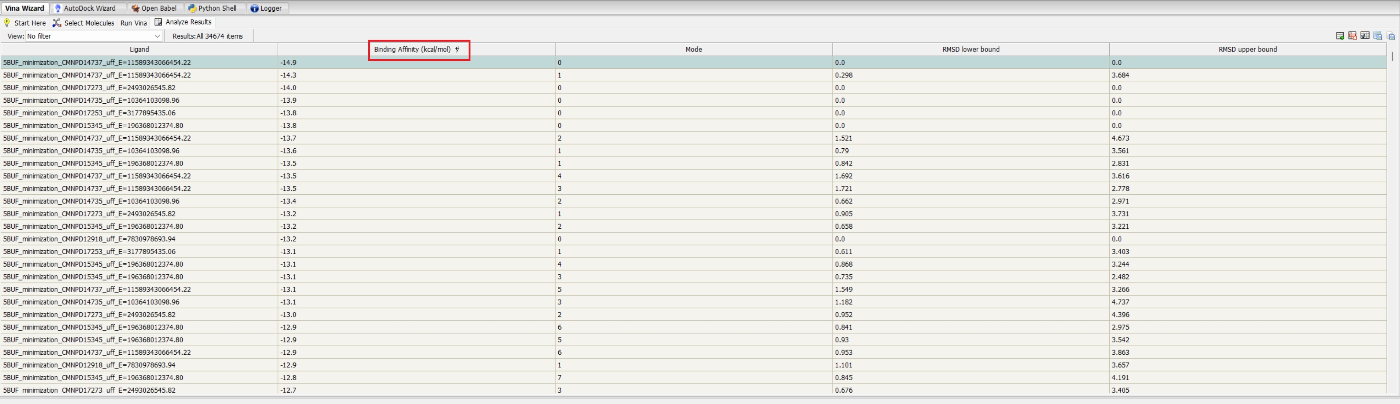
E(エネルギー)が高すぎるものについては、エネルギー最小化のところでバグっているので、取り除きます。
CMNPD14737のもの
実際開いてみるとバグっていることがわかります。

エネルギー計算がうまくいっていないものを取り除くと、
最もbinding affinityが高いのはCMNPD14239で、-7.7 kcal/molになりました。
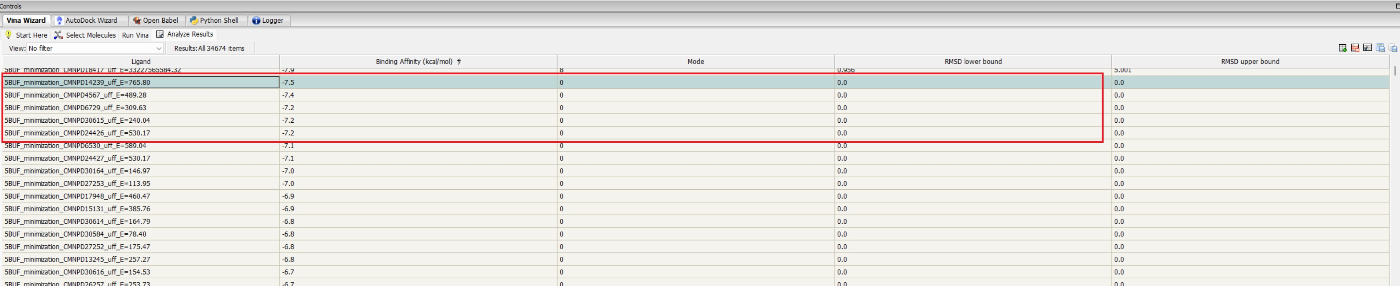
ちなみに、論文で紹介されているCMNPD28986については-5.5kcal/mol(論文では-7.9 kcal/mol)になりました。
選択して、Moleculeタブに行くと、画面に結合を移すことができます。
詳細な解析については他の可視化ツールの方がよいので、そちらで可視化させます。
一番結合力の高いリガンドのPDBファイルを保存してください。

この保存したものとタンパク質ファイルをDiscovery Studioで開いてください。
最初はタンパク質とリガンドは別々のタブで表されますが、各タブの上でCtrl+Cを押し、
以下の赤枠でCtrl + V を押すと一緒に表示できます。
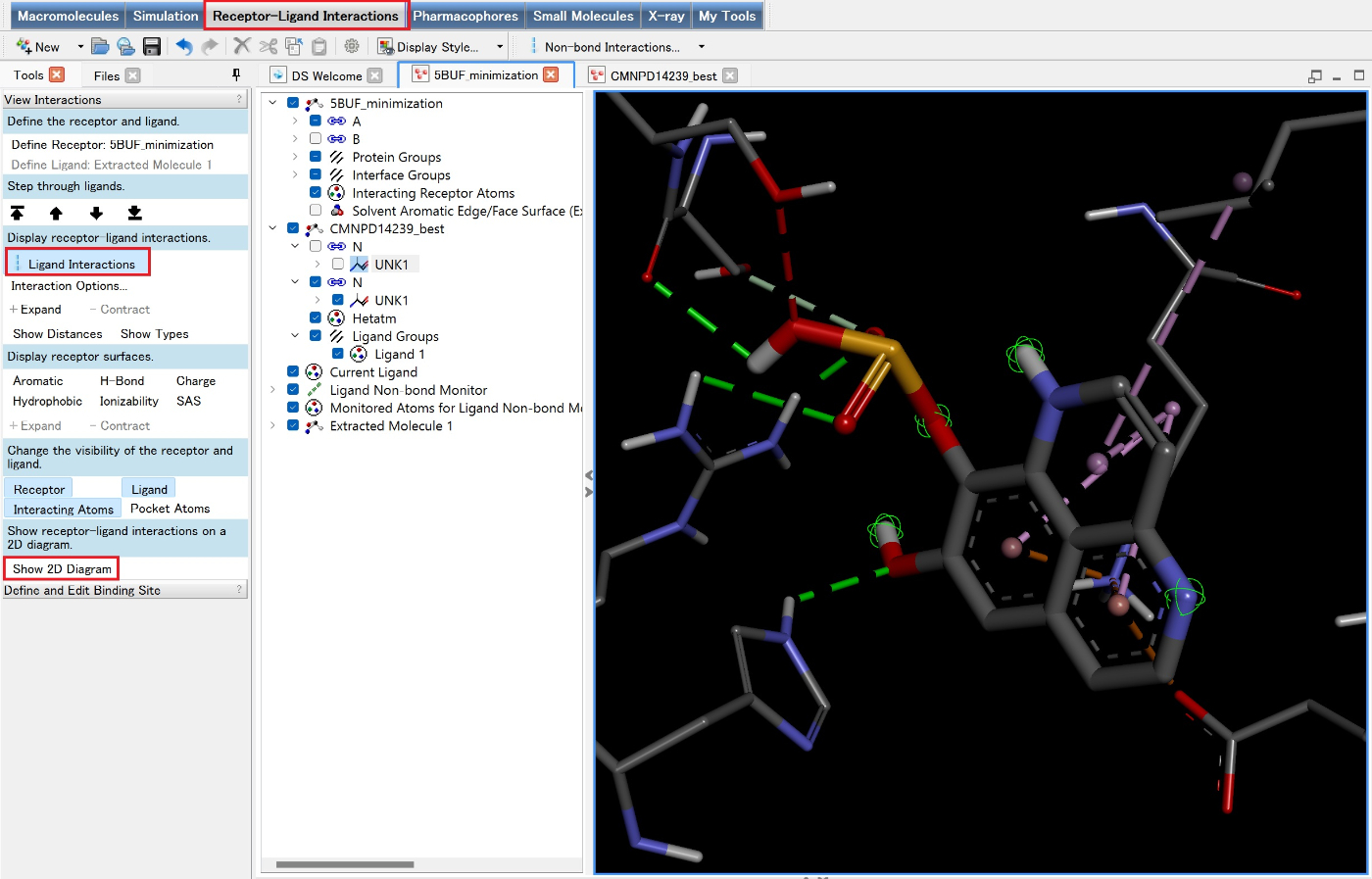
次にReceptor-Ligand Interaction→Ligand Interactionを押すと、以下のような相互作用を表示できます。
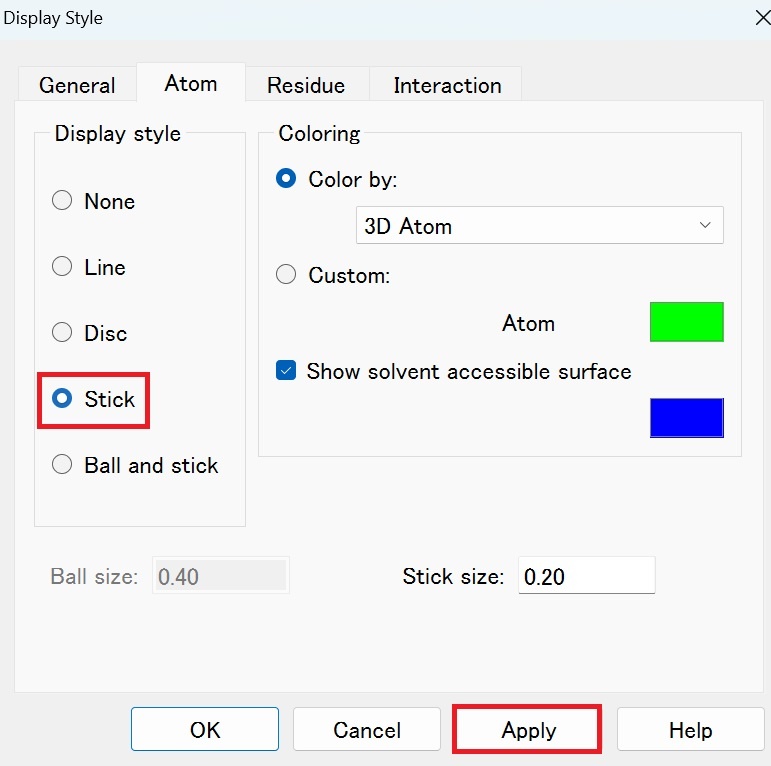
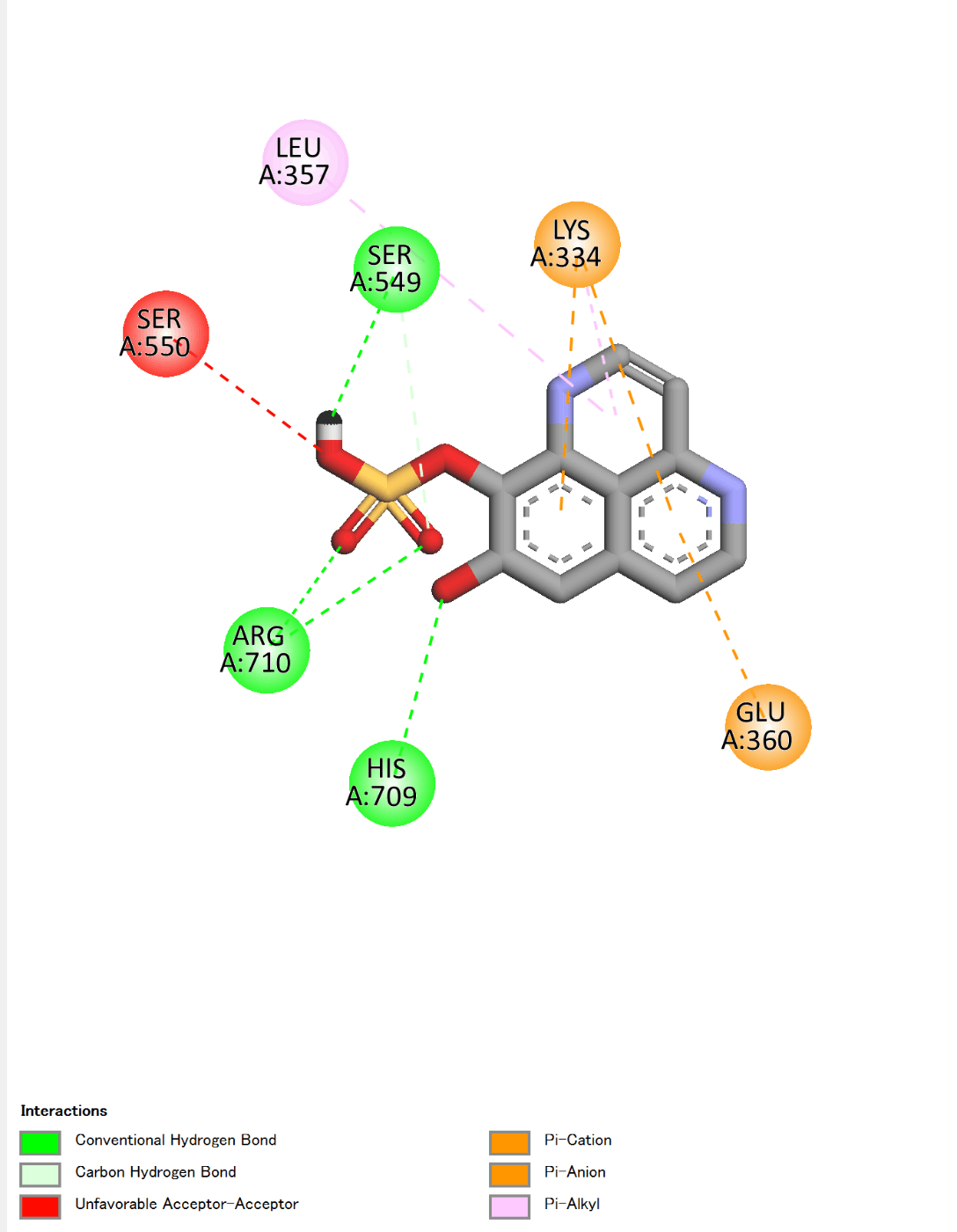
さらにShow 2D Diagramを押すと、以下のように表示できます。

この2D Diagram上で右クリック→Display StyleでAtomをStickにし、Applyをすることで、
論文っぽい図になります。
ちなみに論文で紹介されていたCMNPD28986については以下のようになり、
論文と一緒の結果にはなりませんでした。
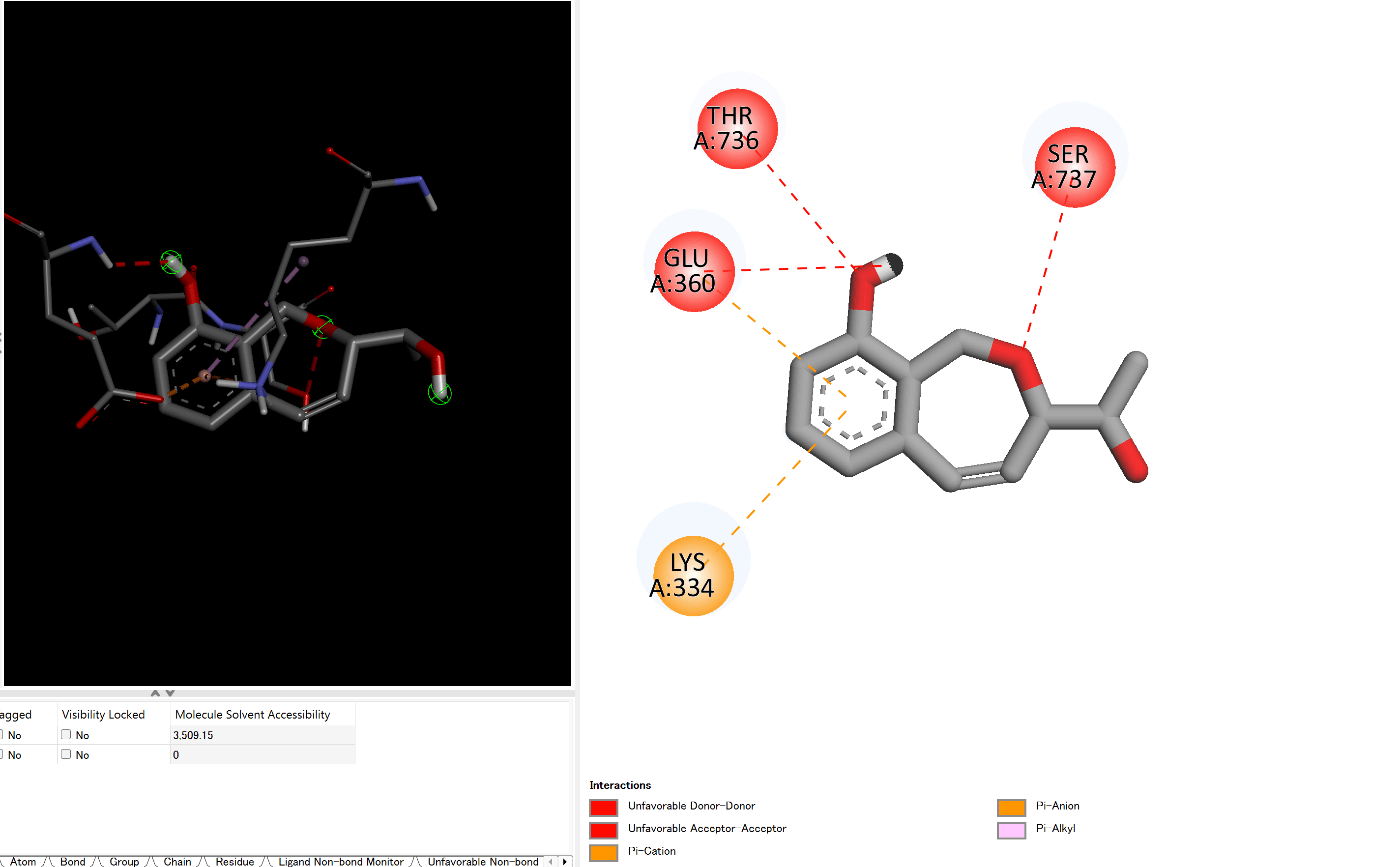
Effectivenessをもっとあげれば、結果が同じになる可能性はあります。しかし、AutoDockは遺伝的 アルゴリズムが用いられているため、論文に書かれていないSeed値が必要だったり、ドッキング環境が異なれば、再現性が難しいらしいので、今回はこの結果を用いて、次のMD simulationに進みます。
最後に
今回は大規模ライブラリからのスクリーニングを行いました。スパコンでなくとも、簡単に大規模ライブラリからのスクリーニングができるのは驚きですね!次は取れてきたリガンドに対して、MD simulationをやっていきます。
参考文献
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
Discussion