GROMACSでリガンド-タンパク質 MDsimulation(分子動力学シミュレーション)を論文に沿ってやってみた
本記事はリガンド-タンパク質 MDsimulation(分子動力学シミュレーション)を論文に沿って、行っています。分子のエネルギーや運動を計算するための相互作用モデルであるフォースフィールドはFf14SBで行なっており、GROMACSのチュートリアルとは異なる方法で行なっています。
エラーの回避方法も含め、詳し目に書いているので、ぜひご覧ください!
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB
本記事は以下の論文のフォローになります。
※本論文のMD simulationはAMBERを用いて行なっておりますが、本記事ではGROMACSでフォローしています。
本論文は標的の選定方法からスクリーニング、スクリーニングした薬物候補化合物の毒性、物性予測、MDシミュレーションまでしており、ほぼ無料のツールですることができます。in silico創薬の基本が詰まっているので、一緒に勉強していきましょう。
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
In silico創薬とその流れ
In silico 創薬、in silicoスクリーニングとは
インシリコ創薬(in silico drug discovery)は、コンピュータを駆使し、シミュレーションやデータ解析を用いて新薬を設計・発見する手法です。
これにより、従来の実験的な方法に比べてコストと時間を大幅に削減できるとされています。
その中でもin silicoスクリーニングは、膨大なデータベースから有望な候補物質を迅速に特定し、その効果や副作用を予測することで、創薬の初期段階での効率化が図られます。
一見難しそうなin silico創薬ですが、現在では様々なアプリケーションやwebサイトがあり、それらを駆使すれば、誰でも簡単に創薬をすることもできます。本記事では、それらのアプリケーションを駆使し、in silico創薬を行った論文をもとに、手法をわかりやすく説明していきます。
In silico創薬の流れ
- ターゲット選定、準備:疾患の原因となる分子(ターゲット)を特定し、その3D構造を準備します。
- 化合物ライブラリの構築:in silicoスクリーニングに使用する化合物集団(=ライブラリ)を作成します。
- in silicoスクリーニング:コンピュータ上で数百万の化合物を対象に、ターゲット分子との結合親和性を評価します。
-
分子動力学シミュレーション(MD simulation):候補化合物とターゲットの相互作用を動的に解析し、安定性や効果を予測します。
4.1.環境構築:Windowsでの環境構築を示しています。
4.2.MD simulation(本記事):MD simulationを行います。結果の可視化を行います。
4.3.RMSD, RMSF, RoG, hydrogen bond, RDF 解析:構造変化や分子間相互作用を評価するための解析手法を行います。
4.4.MM-GBSA解析とMM-PBSA解析、decomposition解析、アラニンスキャン:分子間相互作用や結合エネルギーへの各残基の寄与を解析します。 - 物性、毒性評価:過去のデータを用いて、新たな候補物質の特性や副作用を予測するモデルを構築します。
- (実験的検証):in silico解析で得られた候補物質を実験室で合成し、実際の効果や安全性を検証します。
すでに公開したものについては、リンクを貼っています。今回はMaterial and Methodの「EPSP-inhibitors dynamics analysis」をフォローしています。
各パラメータは以下になります。
力場:AMBER20、Ff14SB(タンパク質)、GAFF(リガンド)
水モデル:TIP3P
エネルギー最小化:1000ステップ
温度:徐々に310K、Langevinダイナミクスで20psの温度上昇
NPT、NVT平衡化:100ps
MD:100ns
論文ではAMBERで行っていますが、今回はGROMACSで行っています。適宜パラメータを変えています。
本記事で用いられるツール一覧
Avogadro: リガンドの水素付加、mol2ファイルへの保存
GROMACS: MDシミュレーション各種設定
acpype: リガンドへの力場付与
全体の流れ
本simulationは以下の流れで行っていきます。
- リガンドの準備
- タンパク質の準備
- リガンドータンパク質の準備
- 水とイオン追加
- エネルギー最小化
- NVT(定温)とNPT(定圧定温)の平衡化
- MDシミュレーション
- 可視化
リガンドの準備
モデルのリガンドはCMNPD28986(TOP-2)です。
まずはAvogadroをインストールしてください。
AvogadroからCMNPD28986を開くと、以下のようになっています。

Edit/Adjust Hydrogensから水素を付加してください。

Exportでligand.mol2としてmol2ファイルで保存してください。
GROMACSではAMBER力場を直接使うことはできないため、リガンドに対してGAFF(General Amber Force Field)を適用するためにACPYPEを使用します。
適当なディレクトリに行き、acpypeのツールをインストールしてください。
git clone https://github.com/alanwilter/acpype.git
次にacpypeディレクトリにligand.mol2を入れます。
以下のディレクトリ構成は以下の感じです。

ligand.mol2の中身をテキストファイルで開いてみてみます。
@<TRIPOS>MOLECULE
CMNPD28986
29 30 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 C 6.9820 26.0100 -21.2750 C.ar 1 UNL1 -0.0198
2 C 7.5840 25.5080 -20.1250 C.ar 1 UNL1 -0.0577
3 C 6.9120 25.5620 -18.9070 C.ar 1 UNL1 -0.0540
4 C 5.6260 26.1050 -18.8120 C.ar 1 UNL1 -0.0164
5 C 5.0030 26.6070 -19.9900 C.ar 1 UNL1 0.0266
6 C 5.6980 26.5570 -21.2150 C.ar 1 UNL1 0.1228
7 C 5.0430 26.1520 -17.4450 C.2 1 UNL1 -0.0556
8 C 3.8820 26.6900 -17.0380 C.2 1 UNL1 -0.0526
9 C 2.8460 27.2920 -17.9410 C.3 1 UNL1 0.1031
10 O 3.4560 28.0730 -18.9240 O.3 1 UNL1 -0.3653
11 C 3.6140 27.1780 -19.9790 C.3 1 UNL1 0.0775
12 C 1.8320 28.1280 -17.1560 C.3 1 UNL1 0.0821
13 C 2.4600 29.3820 -16.5690 C.3 1 UNL1 -0.0362
14 O 1.2960 27.3660 -16.1180 O.3 1 UNL1 -0.3892
15 H 1.3020 26.4290 -16.3680 H 1 UNL1 0.2099
16 O 5.1350 27.0310 -22.3410 O.3 1 UNL1 -0.5064
17 H 4.2130 26.9390 -22.4770 H 1 UNL1 0.2921
18 H 7.5044 25.9769 -22.2082 H 1 UNL1 0.0654
19 H 8.5635 25.0805 -20.1770 H 1 UNL1 0.0619
20 H 7.3877 25.1813 -18.0274 H 1 UNL1 0.0624
21 H 5.6311 25.6927 -16.6781 H 1 UNL1 0.0621
22 H 3.6798 26.6893 -15.9873 H 1 UNL1 0.0599
23 H 2.3158 26.4890 -18.4089 H 1 UNL1 0.0666
24 H 3.4375 27.6892 -20.9022 H 1 UNL1 0.0611
25 H 2.9148 26.3773 -19.8573 H 1 UNL1 0.0611
26 H 1.0654 28.4249 -17.8409 H 1 UNL1 0.0619
27 H 3.2307 29.1059 -15.8800 H 1 UNL1 0.0255
28 H 2.8801 29.9726 -17.3562 H 1 UNL1 0.0255
29 H 1.7112 29.9501 -16.0576 H 1 UNL1 0.0255
@<TRIPOS>BOND
1 16 17 1
2 6 16 1
3 1 6 ar
4 1 2 ar
5 5 6 ar
6 2 3 ar
7 5 11 1
8 4 5 ar
9 10 11 1
10 9 10 1
11 3 4 ar
12 4 7 1
13 9 12 1
14 8 9 1
15 7 8 2
16 12 13 1
17 12 14 1
18 14 15 1
19 1 18 1
20 2 19 1
21 3 20 1
22 7 21 1
23 8 22 1
24 9 23 1
25 11 24 1
26 11 25 1
27 12 26 1
28 13 27 1
29 13 28 1
30 13 29 1
gromacsのチュートリアルによると、この@<TRIPOS>BONDが昇順になっていないとエラーが出るらしいので、以下のスクリプトを sort_mol2_bonds.pl として作って、実行します。
#!/usr/bin/perl
use strict;
# sort_mol2_bonds.pl - a script to reorder the listing in a .mol2 @<TRIPOS>BOND
# section so that the following conventions are preserved:
# 1. Atoms on each line are in increasing order (e.g. 1 2 not 2 1)
# 2. The bonds appear in order of ascending atom number
# 3. For bonds involving the same atom in the first position, the bonds appear
# in order of ascending second atom
#
# Written by: Justin Lemkul (jalemkul@vt.edu)
#
# Distributed under the GPL-3.0 license
unless (scalar(@ARGV)==2)
{
die "Usage: perl sort_mol2_bonds.pl input.mol2 output.mol2\n";
}
my $input = $ARGV[0];
my $output = $ARGV[1];
open(IN, "<$input") || die "Cannot open $input: $!\n";
my @in = <IN>;
close(IN);
# test for header lines that some scripts produce
unless($in[0] =~ /TRIPOS/)
{
die "Nonstandard header found: $in[0]. Please delete header lines until the TRIPOS molecule definition.\n";
}
open(OUT, ">$output") || die "Cannot open $output: $!\n";
# get number of atoms and number of bonds from mol2 file
my @tmp = split(" ", $in[2]);
my $natom = $tmp[0];
my $nbond = $tmp[1];
# check
print "Found $natom atoms in the molecule, with $nbond bonds.\n";
# print out everything up until the bond section
my $i=0;
while (!($in[$i] =~ /BOND/))
{
print OUT $in[$i];
$i++;
}
# print the bond section header line to output
print OUT $in[$i];
$i++;
# read in the bonds and sort them
my $bondfmt = "%6d%6d%6d%5s\n";
my @tmparray;
# sort the bonds - e.g. the one that has the
# lowest atom number in the first position and then the
# lowest atom number in the second position (swap if necessary)
for (my $j=0; $j<$nbond; $j++)
{
my @tmp = split(" ", $in[$i+$j]);
# parse atom numbers
my $ai = $tmp[1];
my $aj = $tmp[2];
# reorder if second atom number < first
if ($aj < $ai)
{
$ai = $tmp[2];
$aj = $tmp[1];
}
# store new lines in a temporary array
$tmparray[$j] = sprintf($bondfmt, $tmp[0], $ai, $aj, $tmp[3]);
}
# loop over tmparray to find each atom number
my $nbond = 0;
for (my $x=1; $x<=$natom; $x++)
{
my @bondarray;
my $ntmp = scalar(@tmparray);
for (my $b=0; $b<$ntmp; $b++)
{
my @tmp = split(" ", $tmparray[$b]);
if ($tmp[1] == $x)
{
push(@bondarray, $tmparray[$b]);
splice(@tmparray, $b, 1);
$ntmp--;
$b--;
}
}
if (scalar(@bondarray) > 0) # some atoms will only appear in $aj, not $ai
{
my $nbondarray = scalar(@bondarray);
if ($nbondarray > 1)
{
# loop over all bonds, find the one with lowest $aj
# and then print it
for (my $y=0; $y<$nbondarray; $y++)
{
my @tmp2 = split(" ", $bondarray[$y]);
my $tmpatom = $tmp[2];
my $lowindex = 0;
if ($tmp2[2] < $tmpatom)
{
$lowindex = $y;
}
my $keep = splice(@bondarray, $lowindex, 1);
$y--;
$nbondarray--;
my @sorted = split(" ", $keep);
$nbond++;
printf OUT $bondfmt, $nbond, $sorted[1], $sorted[2], $sorted[3];
}
}
else
{
$nbond++;
my @tmp2 = split(" ", $bondarray[0]);
printf OUT $bondfmt, $nbond, $tmp2[1], $tmp2[2], $tmp2[3];
}
}
}
close(OUT);
exit;
以下を実行してください。
perl sort_mol2_bonds.pl ligand.mol2 ligand_fix.mol2
続いて、
以下のコードを実行する前にopenBabelをインストールしておいてください。
conda install -c conda-forge openbabel
cd acpype でacpypeディレクトリに行き、以下のコードを実行していきます。
ACPYPEを使ったリガンドの準備:このコマンドは、リガンドのGAFF力場に基づくパラメータファイルを生成します。これにより、GROMACSで使用可能なligand_GMX.groやligand_GMX.topなどのファイルがligand.acpypeに出力されます。
python3 run_acpype.py -i ligand_fix.mol2
上記を実行すると、ligand_fix.acypeに色々なファイルが生成していると思います。

以上で、リガンドの準備は終了です。
タンパク質の準備(Ff14SB force field)
続いて、protein.pdbがあるディレクトリに移動して、以下を実行していきます。
GROMACSでタンパク質をパラメータ化するためにpdb2gmxを使用します。
タンパク質のパラメータ化:
GROMACSのFf14SBに相当するAMBER力場を使うため、以下のコマンドを実行します。GROMACSにはAMBER力場が組み込まれています。このコマンドでは、Ff14SBに相当するamber99sb-ildn力場を指定し、TIP3P水モデルを使用します。
gmx pdb2gmx -f protein.pdb -o protein_processed.gro -ignh
gmx pdb2gmx
gmxはGROMACSのコマンドラインツールのプレフィックスで、pdb2gmxはPDBファイルを読み込み、GROMACSで使用できる形式に変換するコマンドです。具体的には、PDBファイルから力場を選択して、適切なパラメータ(結合情報、水素原子の追加、末端処理など)を設定して出力します。
f protein.pdb
fは「ファイル」を意味するオプションです。後ろに続くprotein.pdbは入力ファイル名で、ここではPDB形式のタンパク質構造ファイルを指定しています。このファイルには、タンパク質の三次元構造情報が記述されています。 o protein_processed.gro
oは「出力ファイル」を指定するオプションです。protein_processed.groは出力されるGROMACS用のファイル名で、.gro形式の座標ファイルとして保存されます。.groファイルは、GROMACSのシミュレーションで使われる標準的なフォーマットで、分子の座標とボックスサイズの情報が含まれています。
ignh
このオプションは「入力ファイル内の水素原子を無視する」ことを意味します。PDBファイルにはしばしば不完全な水素原子の情報が含まれていますが、このオプションを使用することで、既存の水素原子を無視し、GROMACSが必要に応じて自動的に水素原子を追加してくれます。
以下のように出力します。
:-) GROMACS - gmx pdb2gmx, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/protein_ligand_complex_ver2
Command line:
gmx pdb2gmx -f protein.pdb -o protein_processed.gro
Select the Force Field:
From '/usr/local/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
amber99sb-ildn を指定するので、6を選択します。
Using the Amber99sb-ildn force field in directory amber99sb-ildn.ff
Opening force field file /usr/local/gromacs/share/gromacs/top/amber99sb-ildn.ff/watermodels.dat
Select the Water Model:
1: TIP3P TIP 3-point, recommended
2: TIP4P TIP 4-point
3: TIP4P-Ew TIP 4-point optimized with Ewald
4: TIP5P TIP 5-point (see https://gitlab.com/gromacs/gromacs/-/issues/1348 for issues)
5: SPC simple point charge
6: SPC/E extended simple point charge
7: None
tip3p を指定するので、1を選択します。
以下のファイルが出力されればOKです!

タンパク質とリガンド複合体ファイルの合成
リガンドとタンパク質を結合した複合体のgroファイルを生成します。これには手動でprotein_processed.groとligand_fix_GMX.groを結合するか、以下の方法で新しい構造ファイルを作成します。
- タンパク質とリガンドを手動で
complex.groファイルにまとめます。
protein_processed.gro にligand_fix_GMX.gro の値を加え、以下の部分を変更してください。
3-PHOSPHOSHIKIMATE 1-CARBOXYVINYLTRANSFERASE
13194 (←ここを変更)
312THR N 1 2.239 3.120 -2.155
312THR H1 2 2.167 3.093 -2.219
312THR H2 3 2.309 3.173 -2.204
312THR H3 4 2.280 3.039 -2.115
312THR CA 5 2.181 3.204 -2.049
312THR HA 6 2.113 3.147 -2.003
312THR CB 7 2.291 3.246 -1.949
312THR HB 8 2.350 3.312 -1.995
312THR CG2 9 2.242 3.308 -1.818
312THR HG21 10 2.321 3.331 -1.761
312THR HG22 11 2.189 3.390 -1.837
312THR HG23 12 2.185 3.242 -1.769
312THR OG1 13 2.364 3.131 -1.914
312THR HG1 14 2.436 3.155 -1.850
312THR C 15 2.114 3.328 -2.109
312THR O 16 2.141 3.361 -2.224
313GLN N 17 2.030 3.395 -2.030
313GLN H 18 1.996 3.345 -1.950
313GLN CA 19 1.984 3.532 -2.047
313GLN HA 20 2.051 3.583 -2.101
313GLN CB 21 1.850 3.528 -2.122
313GLN HB1 22 1.788 3.468 -2.071
313GLN HB2 23 1.866 3.489 -2.212
313GLN CG 24 1.780 3.663 -2.140
313GLN HG1 25 1.838 3.724 -2.195
313GLN HG2 26 1.762 3.705 -2.051
313GLN CD 27 1.647 3.644 -2.212
313GLN OE1 28 1.540 3.644 -2.152
313GLN NE2 29 1.651 3.613 -2.339
313GLN HE21 30 1.739 3.602 -2.386
313GLN HE22 31 1.566 3.599 -2.390
(中略)
754VAL C13135 2.540 3.968 -3.680
754VAL O13136 2.545 4.034 -3.576
755SER N13137 2.428 3.946 -3.747
755SER H13138 2.432 3.882 -3.824
755SER CA13139 2.298 4.008 -3.718
755SER HA13140 2.308 4.046 -3.626
755SER CB13141 2.267 4.118 -3.820
755SER HB113142 2.179 4.161 -3.800
755SER HB213143 2.266 4.081 -3.913
755SER OG13144 2.364 4.221 -3.818
755SER HG13145 2.340 4.291 -3.886
755SER C13146 2.185 3.905 -3.718
755SER O13147 2.203 3.790 -3.760
756GLN N13148 2.068 3.948 -3.671
756GLN H13149 2.067 4.039 -3.630
756GLN CA13150 1.941 3.875 -3.674
756GLN HA13151 1.954 3.786 -3.718
756GLN CB13152 1.896 3.849 -3.528
756GLN HB113153 1.806 3.807 -3.530
756GLN HB213154 1.892 3.936 -3.480
756GLN CG13155 1.993 3.755 -3.454
756GLN HG113156 2.087 3.785 -3.471
756GLN HG213157 1.981 3.662 -3.488
756GLN CD13158 1.971 3.753 -3.302
756GLN OE113159 1.990 3.850 -3.231
756GLN NE213160 1.945 3.638 -3.243
756GLN HE2113161 1.940 3.554 -3.297
756GLN HE2213162 1.930 3.635 -3.144
756GLN C13163 1.842 3.956 -3.758
756GLN OC113164 1.849 3.934 -3.881
756GLN OC213165 1.808 4.066 -3.709
1 UNL C 1 0.698 2.601 -2.127(←ここから)
1 UNL C1 2 0.758 2.551 -2.013
1 UNL C2 3 0.691 2.556 -1.891
1 UNL C3 4 0.563 2.611 -1.881
1 UNL C4 5 0.500 2.661 -1.999
1 UNL C5 6 0.570 2.656 -2.122
1 UNL C6 7 0.504 2.615 -1.745
1 UNL C7 8 0.388 2.669 -1.704
1 UNL C8 9 0.285 2.729 -1.794
1 UNL O 10 0.346 2.807 -1.892
1 UNL C9 11 0.361 2.718 -1.998
1 UNL C10 12 0.183 2.813 -1.716
1 UNL C11 13 0.246 2.938 -1.657
1 UNL O1 14 0.130 2.737 -1.612
1 UNL H 15 0.130 2.643 -1.637
1 UNL O2 16 0.513 2.703 -2.234
1 UNL H1 17 0.421 2.694 -2.248
1 UNL H2 18 0.750 2.598 -2.221
1 UNL H3 19 0.856 2.508 -2.018
1 UNL H4 20 0.739 2.518 -1.803
1 UNL H5 21 0.563 2.569 -1.668
1 UNL H6 22 0.368 2.669 -1.599
1 UNL H7 23 0.232 2.649 -1.841
1 UNL H8 24 0.344 2.769 -2.090
1 UNL H9 25 0.292 2.638 -1.986
1 UNL H10 26 0.106 2.843 -1.784
1 UNL H11 27 0.323 2.911 -1.588
1 UNL H12 28 0.288 2.997 -1.736
1 UNL H13 29 0.171 2.995 -1.606(←ここまでを変更)
7.38930 10.33800 11.32040
topol.topの変更もしてください。
;
; File 'topol.top' was generated
; By user: unknown (1000)
; On host: DESKTOP-5I5GHRA
; At date: Thu Oct 3 20:12:32 2024
;
; This is a standalone topology file
;
; Created by:
; :-) GROMACS - gmx pdb2gmx, 2024 (-:
;
; Executable: /usr/local/gromacs/bin/gmx
; Data prefix: /usr/local/gromacs
; Working dir: /home/shizuku/protein_ligand_complex_ver3
; Command line:
; gmx pdb2gmx -f protein.pdb -o protein_processed.gro -ignh
; Force field was read from the standard GROMACS share directory.
;
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
; Include chain topologies
#include "topol_Protein_chain_A.itp"
#include "topol_Protein_chain_B.itp"
; Include ligand topology (←ここを追加)
#include "ligand_fix_GMX.itp"
; Include water topology
#include "amber99sb-ildn.ff/tip3p.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
; Include topology for ions
#include "amber99sb-ildn.ff/ions.itp"
[ system ]
; Name
3-PHOSPHOSHIKIMATE 1-CARBOXYVINYLTRANSFERASE
[ molecules ]
; Compound #mols
Protein_chain_A 1
Protein_chain_B 1
UNL 1 (←ここを追加)
これにてタンパク質とリガンドの複合体ファイルの完成です。
水の追加と中和
システムを水溶液中に配置し、必要に応じて中和します。システムに12 Åのバッファ(水ボックスの大きさ)を持つ水分子を追加します。
-
水ボックスの追加:
gmx editconf -f complex.gro -o boxed.gro -c -d 1.2 -bt cubicこのコマンドは12 Åのバッファを設定し、システムを水分子で囲みます。
-
溶媒(水)の追加:
gmx solvate -cp boxed.gro -cs spc216.gro -o solvated.gro -p topol.top -
イオンの追加(中和):
ions.mdpを作成します。これについては、論文内で記述がないので、gromacsチュートリアルから取ってきたものをそのまま使います。
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = cutoff ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; long range electrostatic cut-off
rvdw = 1.0 ; long range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions
ligand_fix_GMX.itp をacpype/ligand_acpypeから取ってきてください。現在のディレクトリに入れてください。
以下を実行してください。
gmx grompp -f ions.mdp -c solvated.gro -p topol.top -o ions.tpr
以下のようなエラーが出るかもしれません。
Fatal error:
Syntax error - File ligand_fix_GMX.itp, line 3
Last line read:
'[ atomtypes ]'
Invalid order for directive atomtypes
ligand_fix_GMX.itpファイルの中身が正しい順序になっていないからだそうです。
このリンクを参考にすると、topol topの一部を、Ligand _fix_GMX.itpから取ってきて、張り付けてください。
以下が修正したtopol.topです。
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
[ atomtypes ](←ここ追加)
;name bond_type mass charge ptype sigma epsilon Amb
ca ca 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
ce ce 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c2 c2 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c3 c3 0.00000 0.00000 A 3.39771e-01 4.51035e-01 ; 1.91 0.1078
os os 0.00000 0.00000 A 3.15610e-01 3.03758e-01 ; 1.77 0.0726
oh oh 0.00000 0.00000 A 3.24287e-01 3.89112e-01 ; 1.82 0.0930
ho ho 0.00000 0.00000 A 5.37925e-02 1.96648e-02 ; 0.30 0.0047
ha ha 0.00000 0.00000 A 2.62548e-01 6.73624e-02 ; 1.47 0.0161
h1 h1 0.00000 0.00000 A 2.42200e-01 8.70272e-02 ; 1.36 0.0208
hc hc 0.00000 0.00000 A 2.60018e-01 8.70272e-02 ; 1.46 0.0208
; Include chain topologies
#include "topol_Protein_chain_A.itp"
#include "topol_Protein_chain_B.itp"
; Include ligand topology
#include "ligand_fix_GMX.itp"
; Include water topology
#include "amber99sb-ildn.ff/tip3p.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
; Include topology for ions
#include "amber99sb-ildn.ff/ions.itp"
[ system ]
; Name
3-PHOSPHOSHIKIMATE 1-CARBOXYVINYLTRANSFERASE
[ molecules ]
; Compound #mols
Protein_chain_A 1
Protein_chain_B 1
UNL 1
SOL 52217
現在のligand_fix_GMX.itpは以下です。
一部は消して、[ moleculetype ]のところはUNLとしています。
(topol.topの[ molecules ]のところと名前を合わせましょう。)
; ligand_fix_GMX.itp created by acpype (v: 2023.11.14) on Thu Oct 3 21:04:33 2024
(ここから)
[ atomtypes ]
;name bond_type mass charge ptype sigma epsilon Amb
ca ca 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
ce ce 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c2 c2 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c3 c3 0.00000 0.00000 A 3.39771e-01 4.51035e-01 ; 1.91 0.1078
os os 0.00000 0.00000 A 3.15610e-01 3.03758e-01 ; 1.77 0.0726
oh oh 0.00000 0.00000 A 3.24287e-01 3.89112e-01 ; 1.82 0.0930
ho ho 0.00000 0.00000 A 5.37925e-02 1.96648e-02 ; 0.30 0.0047
ha ha 0.00000 0.00000 A 2.62548e-01 6.73624e-02 ; 1.47 0.0161
h1 h1 0.00000 0.00000 A 2.42200e-01 8.70272e-02 ; 1.36 0.0208
hc hc 0.00000 0.00000 A 2.60018e-01 8.70272e-02 ; 1.46 0.0208
(ここまでは消去)
[ moleculetype ]
;name nrexcl
UNL 3 (←ここは変更)
[ atoms ]
; nr type resi res atom cgnr charge mass ; qtot bond_type
1 ca 1 UNL C 1 -0.148000 12.01000 ; qtot -0.148
2 ca 1 UNL C1 2 -0.102000 12.01000 ; qtot -0.250
3 ca 1 UNL C2 3 -0.147000 12.01000 ; qtot -0.397
4 ca 1 UNL C3 4 -0.011800 12.01000 ; qtot -0.409
5 ca 1 UNL C4 5 -0.159300 12.01000 ; qtot -0.568
6 ca 1 UNL C5 6 0.127100 12.01000 ; qtot -0.441
7 ce 1 UNL C6 7 -0.108200 12.01000 ; qtot -0.549
8 c2 1 UNL C7 8 -0.176200 12.01000 ; qtot -0.725
9 c3 1 UNL C8 9 0.142300 12.01000 ; qtot -0.583
10 os 1 UNL O 10 -0.411600 16.00000 ; qtot -0.995
11 c3 1 UNL C9 11 0.177700 12.01000 ; qtot -0.817
12 c3 1 UNL C10 12 0.151100 12.01000 ; qtot -0.666
13 c3 1 UNL C11 13 -0.089100 12.01000 ; qtot -0.755
14 oh 1 UNL O1 14 -0.595799 16.00000 ; qtot -1.351
15 ho 1 UNL H 15 0.399000 1.00800 ; qtot -0.952
16 oh 1 UNL O2 16 -0.508100 16.00000 ; qtot -1.460
17 ho 1 UNL H1 17 0.422000 1.00800 ; qtot -1.038
18 ha 1 UNL H2 18 0.151000 1.00800 ; qtot -0.887
19 ha 1 UNL H3 19 0.136000 1.00800 ; qtot -0.751
20 ha 1 UNL H4 20 0.135000 1.00800 ; qtot -0.616
21 ha 1 UNL H5 21 0.126000 1.00800 ; qtot -0.490
22 ha 1 UNL H6 22 0.139000 1.00800 ; qtot -0.351
23 h1 1 UNL H7 23 0.036700 1.00800 ; qtot -0.314
24 h1 1 UNL H8 24 0.055700 1.00800 ; qtot -0.258
25 h1 1 UNL H9 25 0.055700 1.00800 ; qtot -0.203
26 h1 1 UNL H10 26 0.049700 1.00800 ; qtot -0.153
27 hc 1 UNL H11 27 0.051033 1.00800 ; qtot -0.102
28 hc 1 UNL H12 28 0.051033 1.00800 ; qtot -0.051
29 hc 1 UNL H13 29 0.051033 1.00800 ; qtot -0.000
すると以下が実行できるようになります。
gmx grompp -f ions.mdp -c solvated.gro -p topol.top -o ions.tpr
このコマンドは、GROMACSのシミュレーション準備ツールであるgromppを使用して、MDシミュレーションの実行準備を行います。具体的には、指定されたパラメータファイル(ions.mdp)、座標ファイル(solvated.gro)、トポロジーファイル(topol.top)を基に、実行可能なシミュレーションの入力ファイル(ions.tpr)を生成します。
続いて、以下を実行してください。
gmx genion -s ions.tpr -o solvated_ions.gro -p topol.top -pname NA -nname CL -neutral
以下のように表示されます。
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/protein_ligand_complex_ver3
Command line:
gmx genion -s ions.tpr -o solvated_ions.gro -p topol.top -pname NA -nname CL -neutral
Reading file ions.tpr, VERSION 2024 (single precision)
Reading file ions.tpr, VERSION 2024 (single precision)
Will try to add 23 NA ions and 0 CL ions.
Select a continuous group of solvent molecules
Group 0 ( System) has 169845 elements
Group 1 ( Protein) has 13165 elements
Group 2 ( Protein-H) has 6560 elements
Group 3 ( C-alpha) has 890 elements
Group 4 ( Backbone) has 2670 elements
Group 5 ( MainChain) has 3562 elements
Group 6 ( MainChain+Cb) has 4342 elements
Group 7 ( MainChain+H) has 4414 elements
Group 8 ( SideChain) has 8751 elements
Group 9 ( SideChain-H) has 2998 elements
Group 10 ( Prot-Masses) has 13165 elements
Group 11 ( non-Protein) has 156680 elements
Group 12 ( Other) has 29 elements
Group 13 ( UNL) has 29 elements
Group 14 ( Water) has 156651 elements
Group 15 ( SOL) has 156651 elements
Group 16 ( non-Water) has 13194 elements
Select a group:
15を選択してください。
solvated_ions.groファイルが生成されていると思います
これにて、水の追加と中和は終了です。
エネルギー最小化
続いて、プロトコルでは1000ステップのエネルギー最小化を行います。
以下をテキストファイルでコピーして、em.mdpファイルを作成してください。
nstepsの部分を1000ステップに変更します。
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 1000 (←ここを変更) ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.2 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.2 ; long range electrostatic cut-off
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; long range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions
DispCorr = no
em.mdpファイルを現在のディレクトリに置いて、エネルギー最小化を行います。
gmx grompp -f em.mdp -c solvated_ions.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
gmx grompp -f em.mdp -c solvated_ions.gro -p topol.top -o em.tpr
-
gmx grompp: GROMACSの前処理コマンドです。力場のパラメータファイルや構造ファイルを使って、シミュレーションを実行するための入力ファイル(.tprファイル)を作成します。 -
f em.mdp: シミュレーションのパラメータファイル(この場合、エネルギー最小化の設定が記載されたem.mdpファイル)を指定します。 -
c solvated_ions.gro: エネルギー最小化の初期構造として使用するGROMACS形式の座標ファイル(溶媒とイオンが含まれたsolvated_ions.gro)。 -
p topol.top: トポロジーファイル(シミュレーションに使用する分子の構造や結合情報などが記載されたファイル)を指定します。 -
o em.tpr: gromppが作成する出力ファイル。em.tprは、GROMACSがシミュレーションを実行するためのバイナリ形式のファイルです。
gmx mdrun -v -deffnm em
-
gmx mdrun: 実際にシミュレーションを実行するためのコマンドです。gromppで作成された.tprファイルを使用して、エネルギー最小化やMDシミュレーションを実行します。 -
v: 実行中に詳細な出力を表示するオプションです。進行状況をリアルタイムで確認できます。 -
deffnm em: 出力ファイルの接頭辞(デフォルトファイル名)を指定します。この場合、すべての出力ファイル(エネルギー最小化のログ、座標、エネルギーファイルなど)がemという名前で生成されます(例:em.log,em.gro,em.edrなど)
以下のように出力できればOKです。
Steepest Descents did not converge to Fmax < 1000 in 1001 steps.
Potential Energy = -2.6026952e+06
Maximum force = 8.7692803e+03 on atom 6580
Norm of force = 4.0724692e+01
GROMACS reminds you: "Everybody's Good Enough For Some Change" (LIVE)
Steepest Descents did not converge to Fmax < 1000
というメッセージがあるため、GROMACSのエネルギー最小化過程で設定した停止条件に達しなかったことを示しています。
そのため、ステップ数をさらに上げるべきかもしれませんが、今回は論文にそって、1000ステップのままでいきます。
リガンドータンパク質の設定
続いて、以下を実行してください。
gmx make_ndx -f ligand_fix_GMX.gro -o index_ligand_fix_GMX.ndx
このコマンドを使うと、特定の原子や分子のグループ(インデックス)を定義し、後の解析やシミュレーションで使用することができます。
-
gmx make_ndx: GROMACSのインデックス作成・編集ツールを起動します。 -
f ligand_fix_GMX.gro: インデックス作成の対象となる構造ファイル(この場合、ligand_fix_GMX.gro)を指定します。この構造ファイルに基づいて、特定の原子や分子をインデックスとして定義します。 -
o index_ligand_fix_GMX.ndx: 出力するインデックスファイルの名前を指定します。結果として作成されるインデックスファイルはindex_ligand_fix_GMX.ndxという名前になります。
以下のように出力されます。
:-) GROMACS - gmx make_ndx, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/protein_ligand_complex_ver2
Command line:
gmx make_ndx -f ligand_GMX.gro -o index_ligand_GMX.ndx
Reading structure file
Going to read 0 old index file(s)
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
0 System : 29 atoms
1 Other : 29 atoms
2 UNL : 29 atoms
nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups
'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues
't': atom type '|': or 'keep' nr 'splitat' nr 'h': help
'r': residue 'res' nr 'chain' char
"name": group 'case': case sensitive 'q': save and quit
'ri': residue index
0 & ! a H* を入力してください。
-
0 & ! a H*は、「システム全体から水素原子を除いたグループ」を作成するコマンドです。 - 水素原子を除外したインデックスを使って、特定の解析やシミュレーションで水素以外の原子にフォーカスできます。
以下のように出力されます。
Copied index group 0 'System'
Found 14 atoms with name H*
Complemented group: 15 atoms
Merged two groups with AND: 29 15 -> 15
q と打ち、対話から出てください。
続いて、以下を実行してください。
gmx genrestr -f ligand_fix_GMX.gro -n index_ligand_fix_GMX.ndx -o posre_ligand_fix_GMX.itp -fc 1000 1000 1000
このコマンドは、GROMACSで位置拘束(Position Restraint)を作成するために使用されます。位置拘束は、シミュレーション中に特定の原子や分子の位置をある程度固定したい場合に便利です。
-
gmx genrestr:- GROMACSの位置拘束ファイル(
posre.itpファイル)を生成するためのコマンドです。このファイルは、指定された原子や分子の位置を固定(または拘束)し、シミュレーション中の特定の動きを制限するために使用されます。
- GROMACSの位置拘束ファイル(
-
f ligand_fix_GMX.gro:-
ligand_fix_GMX.groという構造ファイルを入力ファイルとして使用します。このファイル内の原子座標が位置拘束の対象となります。
-
-
n index_ligand_fix_GMX.ndx:-
index_ligand_fix_GMX.ndxというインデックスファイルを指定します。このファイルは、拘束をかけたい特定の原子グループを定義します。例えば、特定のリガンドや特定の原子のみを拘束したい場合に、このインデックスファイルを使用します。
-
-
o posre_ligand_GMX.itp:-
posre_ligand_GMX.itpという出力ファイル名を指定します。生成されるファイルは、位置拘束のパラメータが記述された.itpファイルです。このファイルは、後でシミュレーションで使用するトポロジーファイルにインクルードされます。
-
-
fc 1000 1000 1000:-
fcは、**拘束力定数(force constants)**を指定します。この場合、x軸、y軸、z軸の各方向において1000 kJ/mol/nm²の拘束力定数を設定します。この値が大きいほど、原子の位置を強く拘束します。 -
1000 1000 1000は、各軸ごとの拘束力定数を表しており、この例ではすべての方向(x, y, z)で同じ拘束力がかかるように設定されています。
-
以下のように出力されます。
(base) shizuku@DESKTOP-5I5GHRA:~/protein_ligand_complex_ver3$ gmx genrestr -f ligand_fix_GMX.gro -n index_ligand_fix_GMX.ndx -o posre_ligand_GMX.itp -fc 1000 1000 1000
:-) GROMACS - gmx genrestr, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/protein_ligand_complex_ver3
Command line:
gmx genrestr -f ligand_fix_GMX.gro -n index_ligand_fix_GMX.ndx -o posre_ligand_GMX.itp -fc 1000 1000 1000
Reading structure file
Select group to position restrain
Group 0 ( System) has 29 elements
Group 1 ( Other) has 29 elements
Group 2 ( UNL) has 29 elements
Group 3 ( System_&_!H*) has 15 elements
Select a group:
3を選択してください。
続いて、Topol.topに以下のLigand position restraintsを追加してください。
;
; File 'topol.top' was generated
; By user: unknown (1000)
; On host: DESKTOP-5I5GHRA
; At date: Thu Oct 3 20:12:32 2024
;
; This is a standalone topology file
;
; Created by:
; :-) GROMACS - gmx pdb2gmx, 2024 (-:
;
; Executable: /usr/local/gromacs/bin/gmx
; Data prefix: /usr/local/gromacs
; Working dir: /home/shizuku/protein_ligand_complex_ver3
; Command line:
; gmx pdb2gmx -f protein.pdb -o protein_processed.gro -ignh
; Force field was read from the standard GROMACS share directory.
;
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
[ atomtypes ]
;name bond_type mass charge ptype sigma epsilon Amb
ca ca 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
ce ce 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c2 c2 0.00000 0.00000 A 3.31521e-01 4.13379e-01 ; 1.86 0.0988
c3 c3 0.00000 0.00000 A 3.39771e-01 4.51035e-01 ; 1.91 0.1078
os os 0.00000 0.00000 A 3.15610e-01 3.03758e-01 ; 1.77 0.0726
oh oh 0.00000 0.00000 A 3.24287e-01 3.89112e-01 ; 1.82 0.0930
ho ho 0.00000 0.00000 A 5.37925e-02 1.96648e-02 ; 0.30 0.0047
ha ha 0.00000 0.00000 A 2.62548e-01 6.73624e-02 ; 1.47 0.0161
h1 h1 0.00000 0.00000 A 2.42200e-01 8.70272e-02 ; 1.36 0.0208
hc hc 0.00000 0.00000 A 2.60018e-01 8.70272e-02 ; 1.46 0.0208
; Include chain topologies
#include "topol_Protein_chain_A.itp"
#include "topol_Protein_chain_B.itp"
; Include ligand topology
#include "ligand_fix_GMX.itp"
; Ligand position restraints (←ここを追加)
#ifdef POSRES
#include "posre_ligand_fix_GMX.itp"
#endif
; Include water topology
#include "amber99sb-ildn.ff/tip3p.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
; Include topology for ions
#include "amber99sb-ildn.ff/ions.itp"
[ system ]
; Name
3-PHOSPHOSHIKIMATE 1-CARBOXYVINYLTRANSFERASE
[ molecules ]
; Compound #mols
Protein_chain_A 1
Protein_chain_B 1
UNL 1
SOL 52194
NA 23
GROMACSのシミュレーションにおいて、特定の分子(タンパク質、リガンドなど)を動きにくくするために位置拘束を使います。これは、特に平衡化(equilibration)の段階で、特定の部分を安定化させたい場合に有効です。
上記の設定で、タンパク質とリガンドの両方を同時に拘束しています。リガンドとタンパク質を別々に拘束したいときはGROMACSのチュートリアルを参考にしてください。
続いて以下を実行してください。
gmx make_ndx -f em.gro -o index.ndx
gmx make_ndx -f em.gro -o index.ndx は、GROMACSでインデックスファイル(index.ndx)を作成・編集するためのコマンドです。このコマンドを使うことで、特定の分子や原子をグループ化し、そのグループをシミュレーションの解析や操作で使用できるようになります。
Reading structure file
Going to read 0 old index file(s)
Analysing residue names:
There are: 890 Protein residues
There are: 1 Other residues
There are: 52194 Water residues
There are: 23 Ion residues
Analysing Protein...
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
0 System : 169799 atoms
1 Protein : 13165 atoms
2 Protein-H : 6560 atoms
3 C-alpha : 890 atoms
4 Backbone : 2670 atoms
5 MainChain : 3562 atoms
6 MainChain+Cb : 4342 atoms
7 MainChain+H : 4414 atoms
8 SideChain : 8751 atoms
9 SideChain-H : 2998 atoms
10 Prot-Masses : 13165 atoms
11 non-Protein : 156634 atoms
12 Other : 29 atoms
13 UNL : 29 atoms
14 NA : 23 atoms
15 Water : 156582 atoms
16 SOL : 156582 atoms
17 non-Water : 13217 atoms
18 Ion : 23 atoms
19 Water_and_ions : 156605 atoms
nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups
'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues
't': atom type '|': or 'keep' nr 'splitat' nr 'h': help
'r': residue 'res' nr 'chain' char
"name": group 'case': case sensitive 'q': save and quit
'ri': residue index
1 | 13 と書き、Enterを押してください。
以下のように出力されます。
Copied index group 1 'Protein'
Copied index group 13 'UNL'
Merged two groups with OR: 13165 29 -> 13194
q と書いて、実行してください。
この操作により、タンパク質とリガンドが1つのグループとして扱えるようになります。
NVTとNPT平衡化
NVT(定温)とNPT(定圧定温)の平衡化を行います。まずは温度を310 Kに徐々に上げる必要があります。
NVT平衡化(20 ps)
以下のようにnvt.mdpを設定し、現在のディレクトリに入れてください。integrator、nsteps、dt、tc-grps、ref_t、gen_tempを論文用に変更しています。
title = Protein-ligand complex NVT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = sd (←ここを変更); Langevin dynamics (stochastic dynamics)
nsteps = 10000 (←ここを変更); 2 * 10000 = 20 ps
dt = 0.002 (←ここを変更); 2 fs
; Output control
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
nstxout-compressed = 500 ; save coordinates every 1.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds to H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighbor searching and vdW
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 20 ; largely irrelevant with Verlet
rlist = 1.2
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_UNL Water_and_ions (←ここを変更); two coupling groups - more accurate
tau_t = 0.1 ; time constant, in ps
ref_t = 310 (←ここを変更); reference temperature, changed to 310 K for each group
; Pressure coupling
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction is not used for proteins with the C36 additive FF
DispCorr = no
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 310 (←ここを変更) ; temperature for Maxwell distribution changed to 310 K
gen_seed = -1 ; generate a random seed
以下を実行してください。
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr
gmx grompp コマンドは、シミュレーションの入力ファイル(.tpr)を作成するために使用されます。このコマンドでは、エネルギー最小化(em.gro)から得られた構造を元に、NVT平衡化の設定ファイル(nvt.mdp)を読み込んで、必要なすべての情報を含むバイナリ形式の入力ファイル(nvt.tpr)を作成します。
以下を実行して、温度の平衡化を行ってください。
gmx mdrun -deffnm nvt
NPT平衡化(100 ps)
続いて、圧力を平衡化するために、以下のnpt.mdpを作成し、現在のディレクトリに入れてください。integrator、nsteps、dt、tcoupl、tc-grps、tau_t、ref_t、pcouplを論文用に変更しています。
※pcouplの項目において、チュートリアルではBerendsen 法が設定されていますが、GROMACS2021以降ではC-rescale が推奨されているので、こちらを使用します。
npt.mdp
title = Protein-ligand complex NPT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = md (←ここを変更); leap-frog integrator
nsteps = 50000 (←ここを変更); 2 * 50000 = 100 ps
dt = 0.002 (←ここを変更); 2 fs
; Output control
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
nstxout-compressed = 500 ; save coordinates every 1.0 ps
; Bond parameters
continuation = yes ; continuing from NVT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds to H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighbor searching and vdW
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 20 ; largely irrelevant with Verlet
rlist = 1.2
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
rcoulomb = 1.2
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale (←ここを変更); modified Berendsen thermostat
tc-grps = Protein_UNL Water_and_ions (←ここを変更) ; two coupling groups - more accurate
tau_t = 0.1 (←ここを変更); time constant, in ps
ref_t = 310 (←ここを変更); reference temperature, one for each group, in K
; Pressure coupling
pcoupl = C-rescale (←ここを変更); 圧力カップリングにC-rescale法を使用
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction is not used for proteins with the C36 additive FF
DispCorr = no
; Velocity generation
gen_vel = no ; velocity generation off after NVT
以下を実行してください。
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr
このコマンドは、GROMACSでNPT平衡化(定圧・定温アンサンブル)を実行するための準備を行うgromppコマンドです。具体的には、NPTシミュレーションの実行に必要な設定ファイル(npt.tpr)を生成します。
続いて、以下を実行し、圧力の平衡化を行います。
gmx mdrun -deffnm npt
生産MDシミュレーション(100 ns)
生産MDシミュレーションを行います。GPUが設定されていない場合は10日かかるので、状況に応じて変更してください。
md.mdpを以下のように設定してください。nsteps、tc-grps、ref_t、pcouplをGROMACSのチュートリアルから変更しています。
md.mdp
title = Protein-ligand complex MD simulation
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000000 (←ここを変更); 2 * 50000000 = 100000 ps (100 ns)
dt = 0.002 ; 2 fs
; Output control
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; save coordinates every 10.0 ps
; Bond parameters
continuation = yes ; continuing from NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds to H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighbor searching and vdW
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 20 ; largely irrelevant with Verlet
rlist = 1.2 ; neighbor search cutoff in nm
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0 ; start of the switching function for vdW
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
rcoulomb = 1.2 ; cutoff for Coulomb interactions in nm
pme_order = 4 ; cubic interpolation for PME
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_UNL Water_and_ions (←ここを変更); two coupling groups - more accurate
tau_t = 0.1 ; time constant, in ps
ref_t = 310 (←ここを変更); reference temperature, one for each group, in K
; Pressure coupling
pcoupl = C-rescale (←ここを変更); 圧力カップリングにC-rescale法を使用
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction is not used for proteins with the C36 additive FF
DispCorr = no
; Velocity generation
gen_vel = no ; continuing from NPT equilibration
以下を実行してください
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_10.tpr
このコマンドは、GROMACSのコマンドラインツールであるgromppを使用して、MDシミュレーションの入力ファイル(.tprファイル)を生成するためのものです。
以下でMDシミュレーションを開始してください。
gmx mdrun -deffnm md_0_10 -v
終了すると、以下のような出力が出ます。
step 50000000, remaining wall clock time: 0 s
Core t (s) Wall t (s) (%)
Time: 1762898.003 73454.087 2400.0
20h24:14
(ns/day) (hour/ns)
Performance: 117.624 0.204
GROMACS reminds you: "I don't fear death because I don't fear anything I don't understand." (Hedy Lamarr)
もし途中でシミュレーションが終了していた場合は、以下のコードで再開できます。
gmx mdrun -s md_0_10.tpr -cpi md_0_10.cpt -deffnm md_0_10
後処理
周期的な境界条件下で行われるシミュレーションと同様に、分子は "壊れて "表示されたり、ボックスを "飛び跳ね "て行ったり来たりすることがあります。 これを解決するために、タンパク質を再中心化します。
以下のコードを実行してください。
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compact
-
gmx trjconv: これはGROMACSのツールで、軌跡ファイルを変換したり処理するために使われるtrjconvコマンドです。 -
s md_0_10.tpr: これは構造情報が含まれた入力ファイル(.tprファイル)です。このファイルは、軌跡データの処理に必要なトポロジーや他の情報を提供します。 -
f md_0_10.xtc: これはシミュレーションの軌跡ファイル(.xtcファイル)です。シミュレーション中の原子の座標が含まれています。 -
o md_0_10_center.xtc: これは出力ファイルの指定です。変換された軌跡を保存するための新しいファイルで、この場合はmd_0_10_center.xtcとして出力されます。 -
center: 指定した分子やグループの重心をシミュレーションボックスの中心に移動させます。これにより、特定の分子が常にボックスの中心付近に維持されるようになります。 -
pbc mol:pbcは周期境界条件(PBC: Periodic Boundary Conditions)を扱うオプションです。この場合、molは分子単位で周期境界条件を適用し、分子がボックスの外に出た場合も分子全体を元の位置に戻します(分子が切り裂かれるのを防ぎます)。 -
ur compact:urは "unit cell representation" を指定します。このオプションでcompactを指定すると、最もコンパクトな形で分子を配置するようにします。これは視覚化や解析の際に便利です。
以下のように出力されます。
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/protein_ligand_complex_ver3
Command line:
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compact
Note that major changes are planned in future for trjconv, to improve usability and utility.
Will write xtc: Compressed trajectory (portable xdr format): xtc
Reading file md_0_10.tpr, VERSION 2024 (single precision)
Reading file md_0_10.tpr, VERSION 2024 (single precision)
Select group for centering
Group 0 ( System) has 169799 elements
Group 1 ( Protein) has 13165 elements
Group 2 ( Protein-H) has 6560 elements
Group 3 ( C-alpha) has 890 elements
Group 4 ( Backbone) has 2670 elements
Group 5 ( MainChain) has 3562 elements
Group 6 ( MainChain+Cb) has 4342 elements
Group 7 ( MainChain+H) has 4414 elements
Group 8 ( SideChain) has 8751 elements
Group 9 ( SideChain-H) has 2998 elements
Group 10 ( Prot-Masses) has 13165 elements
Group 11 ( non-Protein) has 156634 elements
Group 12 ( Other) has 29 elements
Group 13 ( UNL) has 29 elements
Group 14 ( NA) has 23 elements
Group 15 ( Water) has 156582 elements
Group 16 ( SOL) has 156582 elements
Group 17 ( non-Water) has 13217 elements
Group 18 ( Ion) has 23 elements
Group 19 ( Water_and_ions) has 156605 elements
Select a group:
1を選択してください
Select group for output
Group 0 ( System) has 169799 elements
Group 1 ( Protein) has 13165 elements
Group 2 ( Protein-H) has 6560 elements
Group 3 ( C-alpha) has 890 elements
Group 4 ( Backbone) has 2670 elements
Group 5 ( MainChain) has 3562 elements
Group 6 ( MainChain+Cb) has 4342 elements
Group 7 ( MainChain+H) has 4414 elements
Group 8 ( SideChain) has 8751 elements
Group 9 ( SideChain-H) has 2998 elements
Group 10 ( Prot-Masses) has 13165 elements
Group 11 ( non-Protein) has 156634 elements
Group 12 ( Other) has 29 elements
Group 13 ( UNL) has 29 elements
Group 14 ( NA) has 23 elements
Group 15 ( Water) has 156582 elements
Group 16 ( SOL) has 156582 elements
Group 17 ( non-Water) has 13217 elements
Group 18 ( Ion) has 23 elements
Group 19 ( Water_and_ions) has 156605 elements
Select a group:
0を選択してください。
可視化
pymolを開き、まずはem.groを開いてください。

次にmd_0_10_center.xtc を開いてください。以下のような画面になり、loadを押してください。
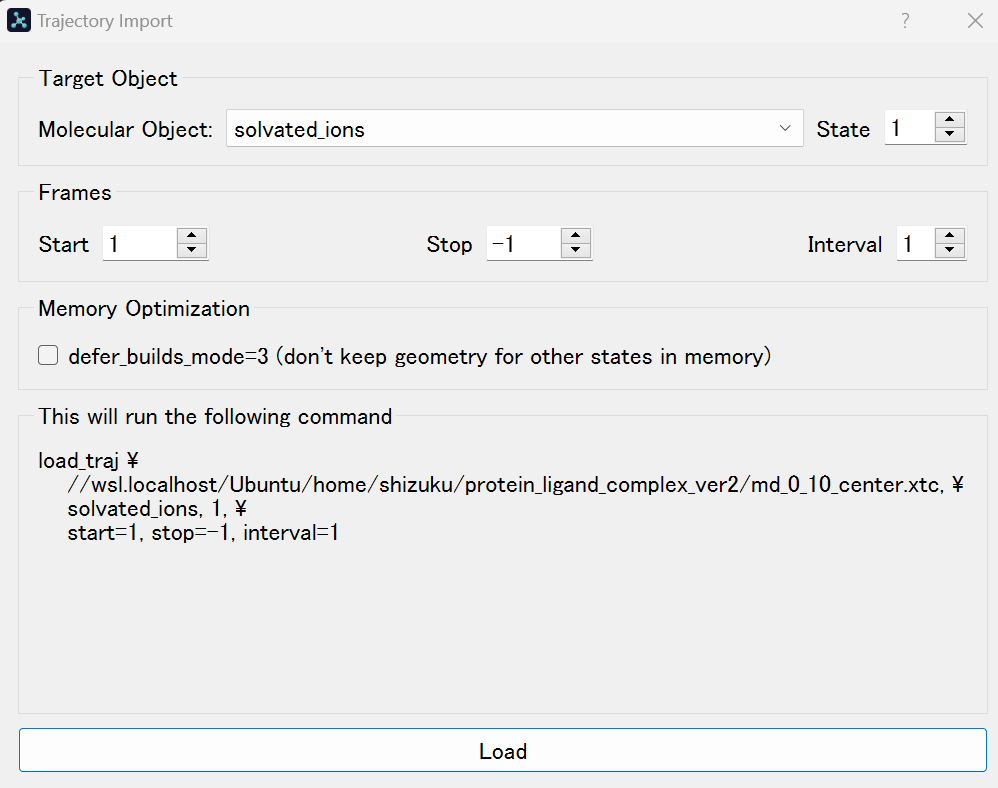
右下の再生ボタンを押すと、simulationが開始されます。
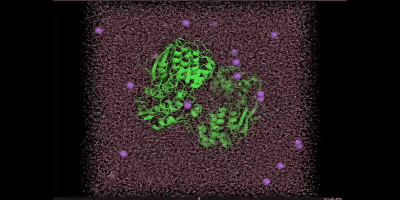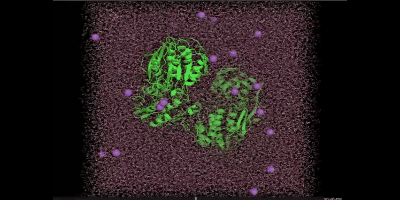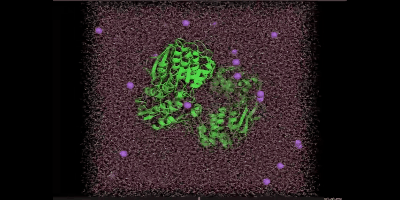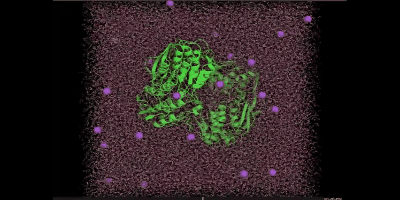
イオンを消したい場合は、下のコマンドで以下のように打ってください。
hide spheres, resn NA+ or resn CL-

水分子も以下で消すことができます。
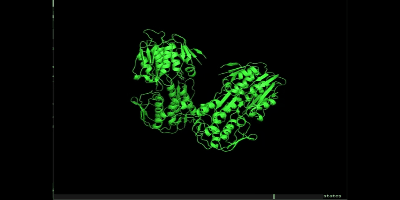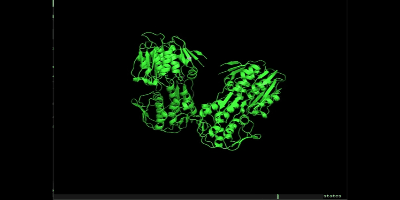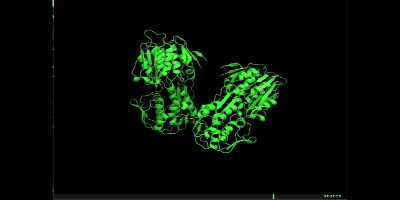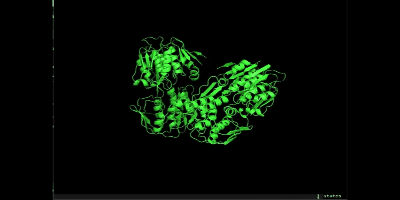
simulationできました!
万が一、シミュレーションでミスっていそうな場合は以下を参照してください。
参考文献
宣伝
本記事を見てくださり、ありがとうございます。
インシリコ創薬についてより学びたい方は
拙著 で学び、さらに色々な方法で新薬探索を楽しんでいただければと思います!
また化合物の評価を行いたい場合は を見ていただければと大変嬉しいです。
Discussion