SnowflakeによるNCBIヒト遺伝子データの取得と取り込み
NCBI遺伝子データの取得と統合:実践ガイド
このガイドでは、NCBI(National Center for Biotechnology Information)から遺伝子データを取得し、Snowflakeに統合する実践的な方法を解説する。実際の検証を通じて、安定したデータパイプラインの構築方法を紹介する。
NCBI遺伝子データの概要
NCBI Geneデータベースは、世界中の生物種の遺伝子情報を包括的に提供している。ヒト(Homo sapiens)の場合、約20万個の遺伝子情報が利用可能である。このデータベースは、遺伝子の基本情報(ID、シンボル、名前)から位置情報(染色体、開始・終了位置)まで、研究に必要な情報を体系的に整理している。
図1: NCBI Geneデータベースのトップページ(https://www.ncbi.nlm.nih.gov/gene/)
この図はNCBI Geneデータベースのトップページである。ここから生物種や遺伝子名で検索し、各遺伝子の詳細情報にアクセスできる。
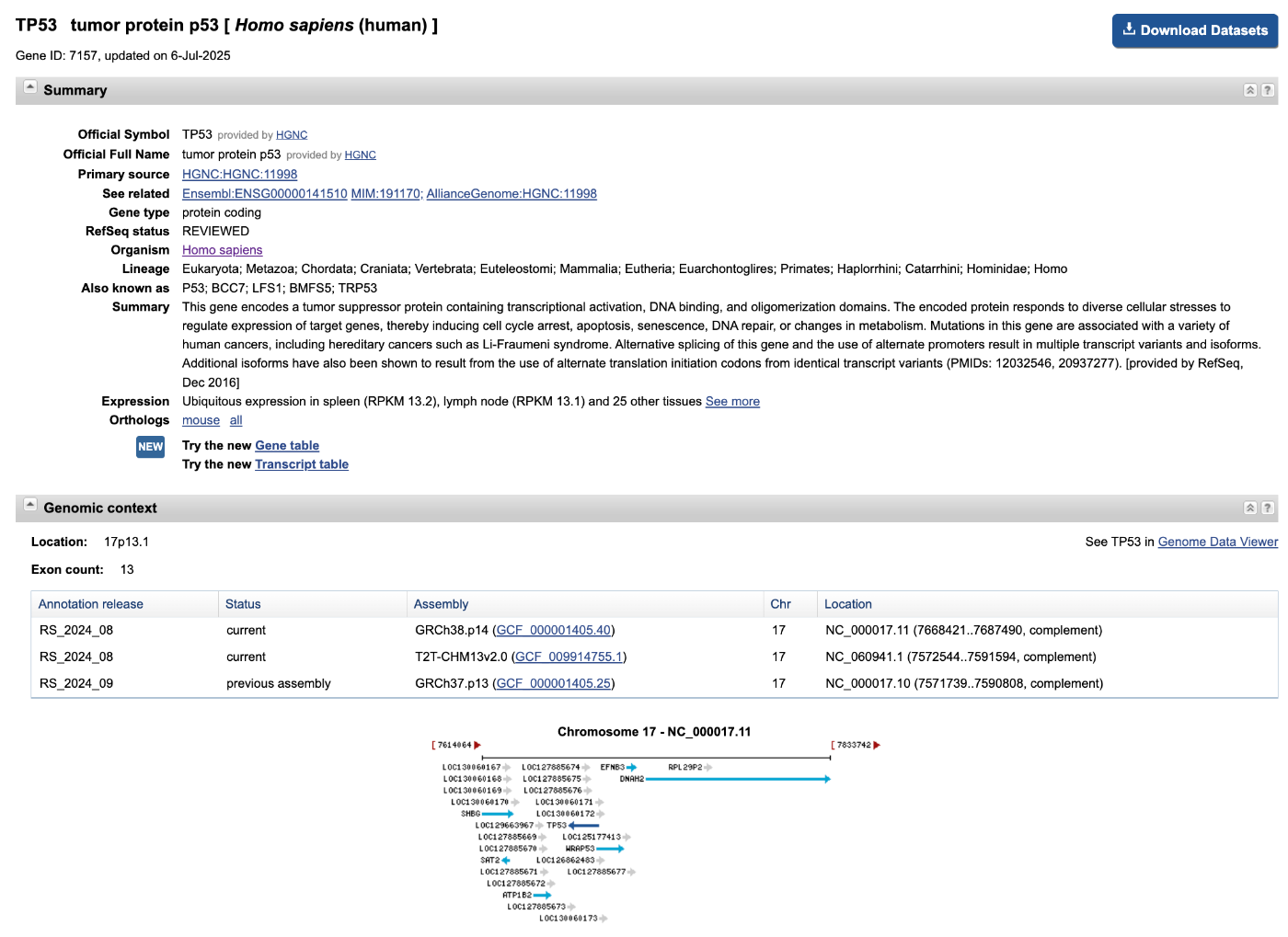
図2: ヒト遺伝子(TP53)の詳細情報ページ例
図2はヒト遺伝子(TP53)の詳細ページの例である。遺伝子シンボル、名称、染色体位置、機能概要など、研究やデータ解析に必要な情報が網羅的に掲載されている。
Snowflake CLIのセットアップ
NCBI遺伝子データをSnowflakeに取り込むのに、Snowflake CLI(コマンドラインインターフェース)のセットアップを行うことにする。CLIを使うことで、スクリプトや自動化パイプラインからSnowflakeを操作できる。
インストール方法
macOSの場合はHomebrew、Linux/Windowsはpipやcondaでインストールできる。
# Homebrew(macOS)
brew install snowflake-cli
# pip
pip install snowflake-cli
# conda
conda install -c conda-forge snowflake-cli
認証情報の設定
Snowflake CLIは~/.snowflake/config.tomlに認証情報を記載する。キーペア認証を推奨する。
[connections.default]
account = "YOUR-ACCOUNT-IDENTIFIER"
user = "YOUR-USERNAME"
private_key_path = "~/.ssh/snowflake_rsa_key.pem"
role = "ACCOUNTADMIN"
warehouse = "BIOINFORMATICS_XS"
authenticator = "SNOWFLAKE_JWT"
接続テスト
snow connection test
「Status OK」と表示されれば接続成功である。(出力イメージ)
(3.9.0) (base) koreeda@dogTK ~/Documents/zenn % snow connection test
+----------------------------------------------------------+
| key | value |
|-----------------+----------------------------------------|
| Connection name | default |
| Status | OK |
| Host | AAAAAA-AB123456.snowflakecomputing.com |
| Account | AAAAAA-AB123456 |
| User | KOREEDA |
| Role | ACCOUNTADMIN |
| Database | not set |
| Warehouse | BIOINFORMATICS_XS |
+----------------------------------------------------------+
Snowflake環境の初期セットアップ
NCBI遺伝子データを格納・解析するには、Snowflake上に専用のデータベースやスキーマ、ウェアハウスを作成しておく必要がある。以下のコマンドをSnowflake CLIで実行しておこう。
1. データベースとスキーマの作成
# バイオインフォマティクス専用データベースの作成
snow sql -q "CREATE DATABASE IF NOT EXISTS BIOINFORMATICS_DB COMMENT = 'バイオインフォマティクス解析用メインデータベース';"
# NCBIデータ用スキーマの作成
snow sql -q "CREATE SCHEMA IF NOT EXISTS BIOINFORMATICS_DB.NCBI_DATA COMMENT = 'NCBI遺伝子データ格納用スキーマ';"
2. ウェアハウスの作成
# 軽量作業用ウェアハウス(メタデータ操作、小規模クエリ)
snow sql -q "CREATE WAREHOUSE IF NOT EXISTS BIOINFORMATICS_XS WITH WAREHOUSE_SIZE = 'X-SMALL' AUTO_SUSPEND = 60 AUTO_RESUME = TRUE INITIALLY_SUSPENDED = TRUE COMMENT = 'メタデータ操作・軽量クエリ用';"
3. 権限の付与(複数ユーザーで利用する場合)
# 研究者ロールの作成と権限付与例
snow sql -q "CREATE ROLE IF NOT EXISTS BIOINFORMATICS_RESEARCHER;"
snow sql -q "GRANT USAGE ON DATABASE BIOINFORMATICS_DB TO ROLE BIOINFORMATICS_RESEARCHER;"
snow sql -q "GRANT USAGE ON SCHEMA BIOINFORMATICS_DB.NCBI_DATA TO ROLE BIOINFORMATICS_RESEARCHER;"
snow sql -q "GRANT USAGE ON WAREHOUSE BIOINFORMATICS_XS TO ROLE BIOINFORMATICS_RESEARCHER;"
これらの初期設定を済ませておくことで、以降のデータ投入や解析がスムーズに進められる。
データ構造
遺伝子データを効率的に管理するため、Snowflakeに適したテーブル構造を設計する。
-- NCBI用データベース・スキーマの作成
CREATE SCHEMA IF NOT EXISTS BIOINFORMATICS_DB.NCBI_DATA;
-- 遺伝子情報テーブル
CREATE TABLE BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO (
gene_id VARCHAR(50),
gene_symbol VARCHAR(100),
gene_name VARCHAR(500),
organism VARCHAR(200),
chromosome VARCHAR(10),
start_position NUMBER,
end_position NUMBER,
strand VARCHAR(1),
gene_type VARCHAR(100),
created_at TIMESTAMP DEFAULT CURRENT_TIMESTAMP()
);
このテーブル構造により、遺伝子の基本情報から位置情報まで、効率的にクエリできるようになる。
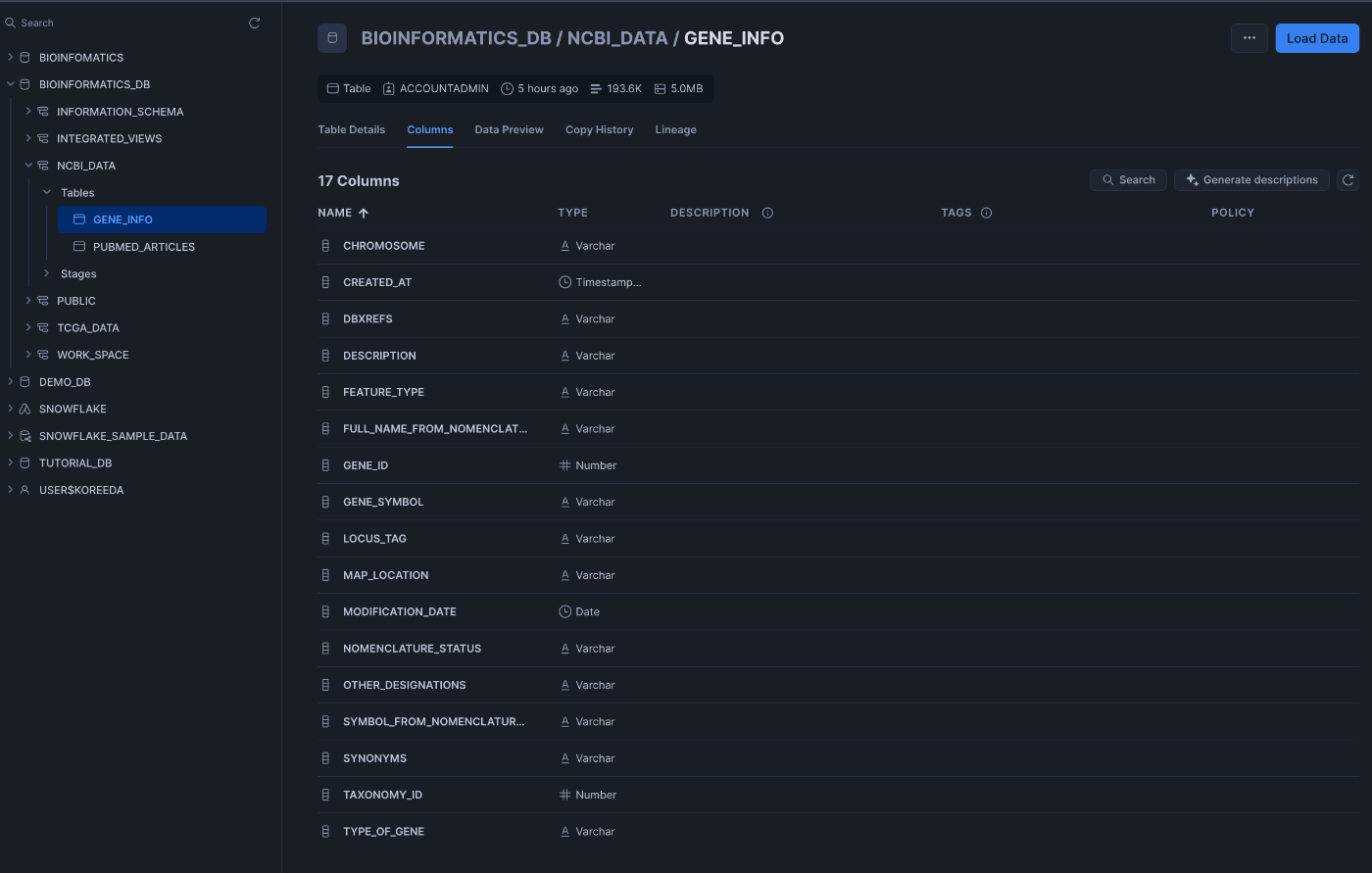
実践的なデータ取得方法
1. FTP一括取得方法
API制限の問題を解決するため、NCBIのFTPサーバーから一括ダウンロードする方法を採用する。この方法により、安定した大量データの取得が可能になる。
#!/bin/bash
# fetch_human_genes_ftp.sh
# 設定
ORGANISM=${1:-"Homo sapiens"}
OUTPUT_FILE="human_genes_$(date +%Y%m%d).csv"
echo "=== NCBI遺伝子データ取得開始 ==="
echo "対象生物種: $ORGANISM"
echo "出力ファイル: $OUTPUT_FILE"
# NCBI FTPサーバーから遺伝子データをダウンロード
echo "1. FTPサーバーからデータをダウンロード中..."
wget -O gene_info.gz "ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_info.gz"
if [ $? -ne 0 ]; then
echo "エラー: FTPダウンロードに失敗しました"
exit 1
fi
# 圧縮ファイルを解凍
echo "2. 圧縮ファイルを解凍中..."
gunzip -f gene_info.gz
if [ $? -ne 0 ]; then
echo "エラー: ファイル解凍に失敗しました"
exit 1
fi
# ヒト遺伝子データを抽出してCSVに変換
echo "3. ヒト遺伝子データを抽出・変換中..."
awk -F'\t' 'BEGIN{OFS=","}
NR==1 {print "gene_id,gene_symbol,gene_name,organism,chromosome,start_position,end_position,strand,gene_type"}
$1 == "9606" { # ヒトのTaxonomy ID
gsub(/,/, ";", $0) # CSVのためにカンマをセミコロンに置換
print $2,$3,$4,$5,$6,$7,$8,$9,$10
}' gene_info > $OUTPUT_FILE
# 一時ファイルを削除
rm -f gene_info
# 統計情報を表示
TOTAL_GENES=$(wc -l < $OUTPUT_FILE)
echo "=== 取得完了 ==="
echo "総遺伝子数: $((TOTAL_GENES - 1))" # ヘッダー行を除く
echo "ファイルサイズ: $(du -h $OUTPUT_FILE | cut -f1)"
echo "出力ファイル: $OUTPUT_FILE"
実行が完了すると、ヒトの全遺伝子情報がCSVファイルとして保存される。このファイルには遺伝子ID、遺伝子シンボル、遺伝子名、染色体位置などの基本情報が含まれている。

2. Snowflakeロード専用スクリプト
CSVファイルをSnowflakeに効率的にロードするためのスクリプトである。ステージの作成からデータロード、品質チェックまで一連の処理を自動化する。
#!/bin/bash
# load_human_genes_to_snowflake.sh
# 設定
CSV_FILE=${1:-"human_genes_$(date +%Y%m%d).csv"}
echo "=== Snowflakeロード開始 ==="
echo "対象ファイル: $CSV_FILE"
# ファイル存在確認
if [ ! -f "$CSV_FILE" ]; then
echo "エラー: ファイル $CSV_FILE が見つかりません"
exit 1
fi
# 既存テーブルを削除(必要に応じて)
echo "1. 既存テーブルを確認中..."
snow sql -q "DROP TABLE IF EXISTS BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO;"
# テーブルを作成
echo "2. テーブルを作成中..."
snow sql -q "
CREATE TABLE BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO (
gene_id VARCHAR(50),
gene_symbol VARCHAR(100),
gene_name VARCHAR(500),
organism VARCHAR(200),
chromosome VARCHAR(10),
start_position NUMBER,
end_position NUMBER,
strand VARCHAR(1),
gene_type VARCHAR(100),
created_at TIMESTAMP DEFAULT CURRENT_TIMESTAMP()
);"
# ステージを作成
echo "3. ステージを作成中..."
snow sql -q "CREATE STAGE IF NOT EXISTS BIOINFORMATICS_DB.NCBI_DATA.GENE_STAGE;"
# ファイルをステージにアップロード
echo "4. ファイルをステージにアップロード中..."
snow sql -q "PUT file://$PWD/$CSV_FILE @BIOINFORMATICS_DB.NCBI_DATA.GENE_STAGE;"
# データをロード
echo "5. データをロード中..."
snow sql -q "
COPY INTO BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO
FROM @BIOINFORMATICS_DB.NCBI_DATA.GENE_STAGE/$CSV_FILE
FILE_FORMAT = (
TYPE = 'CSV'
FIELD_DELIMITER = ','
SKIP_HEADER = 1
FIELD_OPTIONALLY_ENCLOSED_BY = '\"'
NULL_IF = ('', 'NULL', 'null')
);"
# 統計情報を取得
echo "6. ロード結果を確認中..."
snow sql -q "
SELECT
COUNT(*) as total_genes,
COUNT(DISTINCT gene_symbol) as unique_symbols,
COUNT(DISTINCT chromosome) as chromosomes,
MIN(created_at) as first_load,
MAX(created_at) as last_update
FROM BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO;" -o load_results.csv
echo "=== ロード完了 ==="
echo "結果は load_results.csv に保存されました"
ロードが完了すると、遺伝子データがSnowflakeテーブルに格納され、統計情報がCSVファイルに出力される。これにより、データの完全性と品質を確認できる。
3. 一気通貫パイプライン
データ取得からSnowflakeロードまでを自動化する統合スクリプトである。エラーハンドリングとログ出力により、安定した実行が可能になる。
#!/bin/bash
# run_complete_pipeline.sh
echo "=== NCBI遺伝子データ一気通貫パイプライン開始 ==="
echo "開始時刻: $(date)"
# 1. データ取得
echo "ステップ1: データ取得"
./fetch_human_genes_ftp.sh
if [ $? -ne 0 ]; then
echo "エラー: データ取得に失敗しました"
exit 1
fi
# 最新のCSVファイルを取得
CSV_FILE=$(ls -t human_genes_*.csv | head -1)
echo "取得したファイル: $CSV_FILE"
# 2. Snowflakeロード
echo "ステップ2: Snowflakeロード"
./load_human_genes_to_snowflake.sh "$CSV_FILE"
if [ $? -ne 0 ]; then
echo "エラー: Snowflakeロードに失敗しました"
exit 1
fi
echo "=== パイプライン完了 ==="
echo "完了時刻: $(date)"
echo "処理されたファイル: $CSV_FILE"
このスクリプトを実行することで、データ取得からロードまで一連の処理が自動的に実行される。各ステップでエラーが発生した場合は適切に処理が停止し、問題の特定が容易になる。
以下のようにSnowflake上にデータが入っていれば成功である。

Streamlit in Snowflakeによる可視化
ロードしたNCBI遺伝子データを活用して、Streamlit in Snowflakeでインタラクティブな可視化アプリケーションを作成してみよう。これにより、データの探索と分析がより直感的になる。
# gene_visualization_app.py
import streamlit as st
import pandas as pd
import plotly.express as px
import plotly.graph_objects as go
from snowflake.snowpark.context import get_active_session
# ページ設定
st.set_page_config(
page_title="NCBI遺伝子データ可視化",
page_icon="🧬",
layout="wide"
)
st.title("🧬 NCBI遺伝子データ可視化ダッシュボード")
st.markdown("Snowflakeに格納されたヒト遺伝子データの探索と分析")
# Snowflakeセッションの取得
session = get_active_session()
# まずテーブル構造を確認
@st.cache_data
def get_table_structure():
"""テーブル構造を確認"""
query = """
DESCRIBE TABLE BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO
"""
return session.sql(query).collect()
# テーブル構造の表示
st.sidebar.header("📋 テーブル構造")
structure = get_table_structure()
with st.sidebar.expander("カラム情報"):
for row in structure:
st.write(f"**{row['name']}**: {row['type']}")
@st.cache_data
def load_gene_data():
"""遺伝子データを読み込み(実際のカラム名に合わせて修正)"""
query = """
SELECT
GENE_ID,
GENE_SYMBOL,
DESCRIPTION as gene_name,
TAXONOMY_ID,
CHROMOSOME,
MAP_LOCATION,
TYPE_OF_GENE,
SYMBOL_FROM_NOMENCLATURE_AUTHORITY,
FULL_NAME_FROM_NOMENCLATURE_AUTHORITY,
NOMENCLATURE_STATUS,
OTHER_DESIGNATIONS,
MODIFICATION_DATE,
FEATURE_TYPE,
CREATED_AT
FROM BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO
WHERE TAXONOMY_ID = 9606 -- ヒトのTaxonomy ID
AND CHROMOSOME IS NOT NULL
AND CHROMOSOME != ''
"""
return session.sql(query).collect()
@st.cache_data
def get_summary_stats():
"""サマリー統計を取得(実際のカラム名に合わせて修正)"""
query = """
SELECT
COUNT(*) as total_genes,
COUNT(DISTINCT CHROMOSOME) as chromosome_count,
COUNT(DISTINCT TYPE_OF_GENE) as gene_type_count,
COUNT(DISTINCT GENE_SYMBOL) as unique_symbols,
COUNT(DISTINCT TAXONOMY_ID) as organism_count
FROM BIOINFORMATICS_DB.NCBI_DATA.GENE_INFO
WHERE TAXONOMY_ID = 9606 -- ヒトのTaxonomy ID
AND CHROMOSOME IS NOT NULL
AND CHROMOSOME != ''
"""
return session.sql(query).collect()[0]
# データ読み込み
with st.spinner("データを読み込み中..."):
try:
df = load_gene_data()
stats = get_summary_stats()
st.success("データ読み込み完了!")
except Exception as e:
st.error(f"データ読み込みエラー: {str(e)}")
st.info("テーブル構造を確認して、カラム名を修正してください。")
return
# サマリー統計の表示
col1, col2, col3, col4 = st.columns(4)
with col1:
st.metric("総遺伝子数", f"{stats['TOTAL_GENES']:,}")
with col2:
st.metric("染色体数", stats['CHROMOSOME_COUNT'])
with col3:
st.metric("遺伝子タイプ数", stats['GENE_TYPE_COUNT'])
with col4:
st.metric("ユニークシンボル数", f"{stats['UNIQUE_SYMBOLS']:,}")
st.divider()
# タブによる可視化
tab1, tab2, tab3, tab4 = st.tabs(["📊 染色体別分布", "🔍 遺伝子タイプ分析", "📈 遺伝子情報", "🗺️ 染色体マップ"])
with tab1:
st.header("染色体別遺伝子分布")
# 染色体別遺伝子数
chr_counts = pd.DataFrame(df).groupby('CHROMOSOME').size().reset_index(name='count')
chr_counts = chr_counts.sort_values('CHROMOSOME')
fig1 = px.bar(
chr_counts,
x='CHROMOSOME',
y='count',
title="染色体別遺伝子数",
labels={'count': '遺伝子数', 'CHROMOSOME': '染色体'}
)
fig1.update_layout(height=500)
st.plotly_chart(fig1, use_container_width=True)
# 染色体別の詳細統計
chr_stats = pd.DataFrame(df).groupby('CHROMOSOME').agg({
'GENE_ID': 'count',
'GENE_SYMBOL': 'nunique',
'TYPE_OF_GENE': 'nunique'
}).round(2)
chr_stats.columns = ['総遺伝子数', 'ユニークシンボル数', '遺伝子タイプ数']
st.dataframe(chr_stats, use_container_width=True)
with tab2:
st.header("遺伝子タイプ分析")
# 遺伝子タイプ別分布
type_counts = pd.DataFrame(df).groupby('TYPE_OF_GENE').size().reset_index(name='count')
type_counts = type_counts.sort_values('count', ascending=False).head(15)
fig2 = px.pie(
type_counts,
values='count',
names='TYPE_OF_GENE',
title="遺伝子タイプ別分布(上位15種)"
)
fig2.update_layout(height=600)
st.plotly_chart(fig2, use_container_width=True)
# 遺伝子タイプ別の詳細統計
type_stats = pd.DataFrame(df).groupby('TYPE_OF_GENE').agg({
'GENE_ID': 'count',
'GENE_SYMBOL': 'nunique',
'CHROMOSOME': 'nunique'
}).round(2)
type_stats.columns = ['総遺伝子数', 'ユニークシンボル数', '染色体数']
type_stats = type_stats.sort_values('総遺伝子数', ascending=False).head(10)
st.dataframe(type_stats, use_container_width=True)
with tab3:
st.header("遺伝子情報分析")
# 命名法ステータス別分布
nomenclature_counts = pd.DataFrame(df).groupby('NOMENCLATURE_STATUS').size().reset_index(name='count')
nomenclature_counts = nomenclature_counts.sort_values('count', ascending=False)
fig3 = px.bar(
nomenclature_counts,
x='NOMENCLATURE_STATUS',
y='count',
title="命名法ステータス別分布",
labels={'count': '遺伝子数', 'NOMENCLATURE_STATUS': '命名法ステータス'}
)
fig3.update_layout(height=500)
st.plotly_chart(fig3, use_container_width=True)
# 統計情報
col1, col2 = st.columns(2)
with col1:
st.subheader("命名法ステータス統計")
st.dataframe(nomenclature_counts, use_container_width=True)
with col2:
st.subheader("最新更新遺伝子 TOP 10")
# DataFrame化直後にMODIFICATION_DATEを日付型に変換
df = pd.DataFrame(df)
df['MODIFICATION_DATE'] = pd.to_datetime(df['MODIFICATION_DATE'], errors='coerce')
# 最新更新遺伝子 TOP 10
recent_genes = df.nlargest(10, 'MODIFICATION_DATE')[['GENE_SYMBOL', 'gene_name', 'MODIFICATION_DATE', 'CHROMOSOME']]
st.dataframe(recent_genes, use_container_width=True)
with tab4:
st.header("染色体マップ")
# 染色体選択
selected_chr = st.selectbox(
"染色体を選択",
sorted(pd.DataFrame(df)['CHROMOSOME'].unique())
)
if selected_chr:
chr_data = pd.DataFrame(df)[pd.DataFrame(df)['CHROMOSOME'] == selected_chr]
# 染色体上の遺伝子分布(MAP_LOCATIONを使用)
# MAP_LOCATIONから位置情報を抽出する処理を追加
chr_data_with_position = chr_data.copy()
# 簡単な散布図(遺伝子ID vs 遺伝子タイプ)
fig4 = px.scatter(
chr_data_with_position,
x='GENE_ID',
y='TYPE_OF_GENE',
color='NOMENCLATURE_STATUS',
hover_data=['GENE_SYMBOL', 'DESCRIPTION'],
title=f"染色体 {selected_chr} の遺伝子分布",
labels={'GENE_ID': '遺伝子ID', 'TYPE_OF_GENE': '遺伝子タイプ'}
)
fig4.update_layout(height=600)
st.plotly_chart(fig4, use_container_width=True)
# 選択された染色体の統計
st.subheader(f"染色体 {selected_chr} の詳細")
chr_detail = chr_data.agg({
'GENE_ID': 'count',
'GENE_SYMBOL': 'nunique',
'TYPE_OF_GENE': 'nunique',
'NOMENCLATURE_STATUS': 'nunique'
}).round(2)
chr_detail.index = ['総遺伝子数', 'ユニークシンボル数', '遺伝子タイプ数', '命名法ステータス数']
st.dataframe(chr_detail, use_container_width=True)
# サイドバーでの検索機能
st.sidebar.header("🔍 遺伝子検索")
search_term = st.sidebar.text_input("遺伝子シンボルまたは名前で検索")
if search_term:
search_results = pd.DataFrame(df)[
pd.DataFrame(df)['GENE_SYMBOL'].str.contains(search_term, case=False, na=False)].head(20)
if not search_results.empty:
st.sidebar.subheader("検索結果")
for _, row in search_results.iterrows():
with st.sidebar.expander(f"{row['GENE_SYMBOL']}"):
st.write(f"**遺伝子ID:** {row['GENE_ID']}")
st.write(f"**染色体:** {row['CHROMOSOME']}")
st.write(f"**マップ位置:** {row['MAP_LOCATION']}")
st.write(f"**遺伝子タイプ:** {row['TYPE_OF_GENE']}")
st.write(f"**命名法ステータス:** {row['NOMENCLATURE_STATUS']}")
st.write(f"**更新日:** {row['MODIFICATION_DATE']}")
else:
st.sidebar.write("該当する遺伝子が見つかりませんでした。")
# フッター
st.divider()
st.markdown("""
---
**データソース:** NCBI Gene Database
**更新日:** 最新のNCBIデータを使用
**技術スタック:** Snowflake + Streamlit + Plotly
""")
すると下記のようなダッシュボードができます。
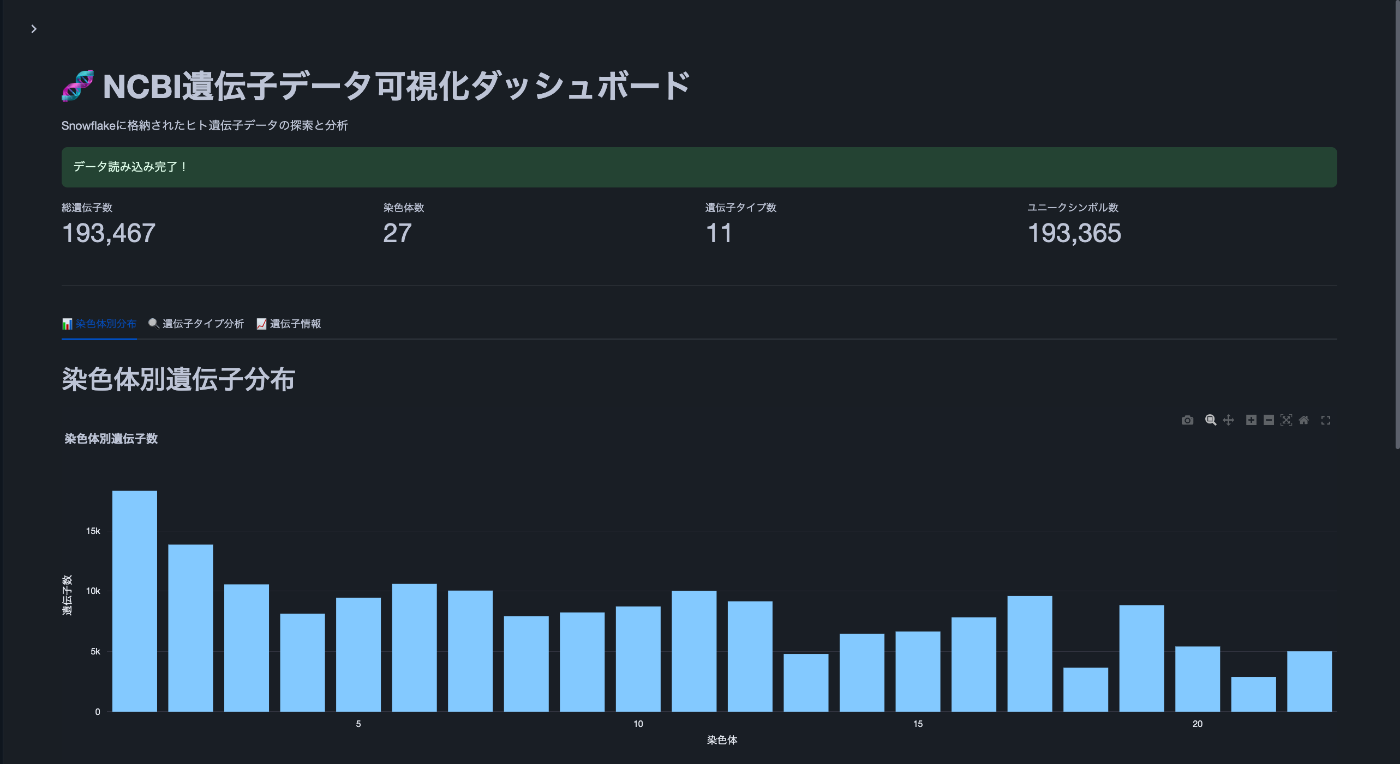
図:染色体別遺伝子数の棒グラフ
各染色体に存在する遺伝子数を示しています。1番染色体や2番染色体など、大きな染色体ほど遺伝子数が多い傾向が見られます。染色体ごとの遺伝子分布の偏りや特徴を俯瞰できます。

図:遺伝子タイプ別分布の円グラフ
遺伝子タイプ(例:protein-coding, ncRNA, pseudogeneなど)ごとの割合を示しています。機能や特徴ごとの遺伝子の多様性を視覚的に理解できます。

表:最新更新遺伝子TOP10
データベースで最近更新・修正された遺伝子情報の上位10件を表示しています。最新の研究動向や注目されている遺伝子を素早く把握するのに役立ちます。
終わりに
ヒトにおいて染色体1が一番遺伝子数多いことや、Protein codingとncRNAが同じくらいの割合で存在していることは私も知らなかったため、可視化してみてとても良い気付きとなった。実際に遺伝子リストを持っていると後続の解析とリスト突合したりできるのでマスターデータとして所有しておくと便利だろう。
宣伝
公共データを用いたシングルセルダイナミクス解析 Trajectory解析、RNA Velocity解析、空間トランスクリプトーム解析のやり方
大人気シリーズ「公共データを用いた〇〇」の第3弾!
Trajectory解析、RNA Velocity解析、空間トランスクリプトーム解析といったシングルセルダイナミクス解析に挑戦しませんか?本書は、シングルセルダイナミクス解析の実践的な解説書として、Trajectory解析、RNA Velocity解析、そして空間トランスクリプトーム解析といった、より高度な解析手法に焦点を当てています。
公共データを用いたRNA-seq解析【Rデータ解析編】 ~Rで始めるドライ研究~
大人気シリーズ「公共データを用いた〇〇」の第4弾!
Rを使ってRNA-seq解析に挑戦しませんか?本書は、R言語を使った実践的なRNA-seq解析の技術書です。プログラミング初心者でも公共データを用いてRNA-seq解析を行い、論文図表作成まで行える内容を提供します!
Discussion