【シングルセル解析】Seurat plot機能のtips
シングルセルデータ解析ツールのSeuratには多彩なplotが用意されているが、各plotに用意されているオプションでは不十分に感じることがある。ここではSeurat plotをより自由に表現するtipsを紹介する。
library(Seurat)
library(dplyr)
library(ggplot2)
library(patchwork)
library(pheatmap)
library(RColorBrewer)
library(viridis)
デモデータ用意
データの準備
PBMC 1万細胞、PBMC 2700細胞のデータを使用する。
- 10X GENOMICSが提供しているPBMC 1万細胞のデータ
https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_10k_v3?
上記ページのFeature/cell matrix HDF5(filtered)のリンクからh5ファイルをダウンロード。
mat <- Read10X_h5(filename = "pbmc_10k_v3_filtered_feature_bc_matrix.h5")
sc <- CreateSeuratObject(mat, min.cells = 3, min.features = 200)
sc <- PercentageFeatureSet(sc, pattern = "^MT-", col.name = "Percent_mito")
sc <- sc %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA() %>%
RunUMAP(dims = 1:30) %>%
FindNeighbors(dims = 1:30) %>%
FindClusters(resolution = 0.5)
# resolution 0.5の時のcluster情報をmeta.dataスロットに保存
sc$cluster0.5 <- sc@active.ident
# cluster数が少ない版も保存しておく。
sc <- FindClusters(sc, resolution = 0.1)
sc$cluster0.1 <- sc@active.ident
- Seuratチュートリアルページで使用されているPBMC 2700細胞のデータ
上記ページのhereのリンクからデーが入ったフォルダをダウンロード、解凍しておく。
pbmc.data <- Read10X(data.dir = "pbmc3k_filtered_gene_bc_matrices/filtered_gene_bc_matrices/hg19")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc <- pbmc %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA() %>%
RunUMAP(dims = 1:10) %>%
FindNeighbors(dims = 1:10) %>%
FindClusters(resolution = 0.5)
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
pbmc$Cell_type <- pbmc@active.ident
【Seuratの修飾コマンド】
軸、凡例の削除
Serutaには凡例や軸を無くすような簡単な修飾コマンドが用意されている。
p1 <- DimPlot(sc)
p2 <- DimPlot(sc) + NoLegend() # 凡例を無くす
p3 <- DimPlot(sc) + NoLegend() + NoAxes() # さらに軸を無くす
p1|p2|p3

NoAxes()にはkeep.text=、keep.ticks=オプションがあり、TRUEにすると、軸ラベルやticksを残すこともできる。
Fontsize
FontSize()機能で、軸のラベルやタイトルのサイズを指定できる。plotのmain titleがある場合はmain=〇〇でサイズ指可能。
DimPlot(sc) +
FontSize(x.text = 4, y.text = 8, x.title = 12, y.title = 16)

軸ラベルを傾ける
x軸ラベルを45度傾ける。RotatedAxis()
DotPlot(sc,
features = c("CD3E", "CD4", "CD8A", "MS4A1", "CD79B", "CD14", "CD68")
) +
RotatedAxis()

【ggplotの修飾コマンド】
Seuratでは上記のように簡単なplot修飾コマンドを用意してくれているが、それでは不十分であったり、Seurat版のコマンドを探すのが面倒だったりする。
基本はggplot2パッケージを使ったplotなので、ggplotの修飾コマンドが機能する。
以下は適当な例だが、ggplotの記法を使えばplotの各要素 (線やtext, legend,,,)などが自由度高く編集可能である。
DimPlot(sc) +
theme(axis.title = element_text(size = 10, face = "bold"),
axis.text.x = element_text(size = 6, colour = "red"),
axis.text.y = element_text(size = 6, colour = "blue"),
axis.line = element_blank(),
axis.ticks = element_line(colour = "gray"),
legend.position = "bottom") +
ggtitle("UMAP")

以降はSeurat plotをggplotで修飾する例を紹介していく。
【離散値の色 (VlnPlotやDimPlot)】
デフォルトの色
DimPlot(sc, group.by = "cluster0.1", label = T)
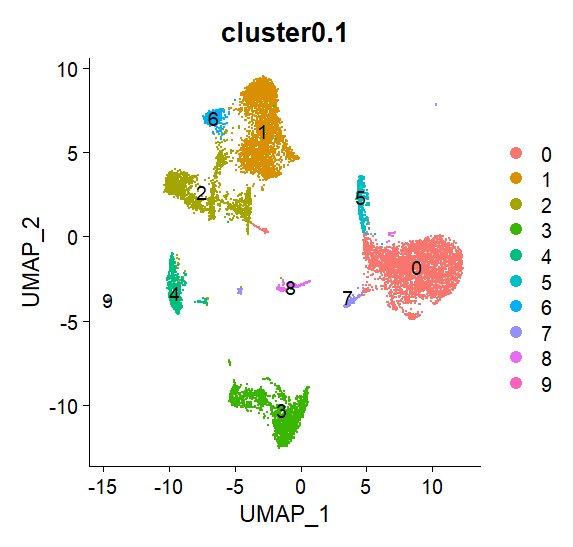
各クラスターの色を変更するにはDimPlotのcols=引数で色を指定する。group.by=引数で指定した情報で群分けした場合の群の数だけ色の指定が必要。色数が足りないとエラーになる。
DimPlot(sc,
group.by = "cluster0.1",
label = T,
cols = rainbow(10))

ggplotの修飾コマンドも使用可能。DimPlot()ではscale_color_シリーズ、VlnPlot()ではscale_fill_シリーズを使用する。
DimPlot(sc,
group.by = "cluster0.1",
label = T) +
scale_color_brewer(palette = "Set3")

DimPlot(sc,
label = T) +
scale_color_viridis_d(option = "turbo")

色ベクトルの用意に関しては、私のスクラップにまとめているのでそちらを参照して欲しい。
【連続値の色 (FeaturePlotやDotPlot, DoHeatmap)】
FeaturePlotの色
遺伝子発現量を可視化するFeaturePlot()にもcols=引数があるが、困ったことにcolsの色数が増えると凡例に表示される色の範囲がおかしくなる。正規化して対数変換をした遺伝子発現量をplotしているのでこの値が変わるのはよろしくない。
FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3)

FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3,
cols = c("lightgray", "darkred"))

FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3,
cols = c("lightgray", "gray", "orange", "red", "darkred", "black"))

発現値の上限が6に上がっている
これを解決するには、ggplotによる色修飾機能を使用する。
FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3) &
scale_color_gradientn(colours = c("lightgray", "gray", "orange", "red", "darkred", "black"))

FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3) &
scale_color_gradientn(colours = rev(brewer.pal(n = 11, name = "RdBu")))

viridisのパレットもscale_color_viridis()機能から使用可能。(※ scale_color_brewer()でRColorBrewerのパレットが使えるが、これは離散値用)
FeaturePlot(sc, features = c("CD3D", "MS4A1", "CD14"), ncol = 3) &
scale_color_viridis_c(option = "D")

DotPlotの色
DotPlot()では、複数色指定してもAverage Expressionの値が変わってしまうことはないが、指定した色ベクトルの最初の2色しか使われてない。
DotPlot(sc, features = c("CD3D", "MS4A1", "CD14"))

DotPlot(sc, features = c("CD3D", "MS4A1", "CD14"),
cols = c("lightgray", "orange", "red", "darkred", "black"))

DotPlotもggplotの色修飾機能で変色可能。
DotPlot(sc, features = c("CD3D", "MS4A1", "CD14")) &
scale_color_gradientn(colours = c("lightgray", "orange", "red", "darkred", "black"))

DotPlot(sc, features = c("CD3D", "MS4A1", "CD14")) &
scale_color_viridis()

DoHeatmapの色
デフォルト色。
DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14"))

DoHeatmap()機能にはそもそも色を指定するオプションが無いが、ggplotの色修飾で編集可能である。なお、FeaturePlot()/DotPlot()ではscale_color_シリーズであったが、DoHeatmap()ではscale_fill_シリーズを使用する。
DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14")) +
scale_fill_gradient(low = "grey", high = "red")

DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14")) +
scale_fill_gradient2(low = "blue",mid = "white",high = "red", midpoint=0)

DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14")) +
scale_fill_gradientn(colors = RColorBrewer::brewer.pal(7, "RdYlBu"))

【値の範囲】
外れ値のような発現を持つ細胞がいるとそっちに色の最大値/最小値が引っ張られてしまい、本来の陽性細胞がぼんやり表現されてしまうことがある。
色を割り当てる値の範囲を調整して改善する例を紹介する。
FeaturePlotの値の範囲
以下の例だと最大発現値を持つ細胞はわずかで、その他陽性細胞はぼんやりしており、どこまでが発現細胞なのか見づらい。そして高発現細胞がどこにいるのかわかりづらい。
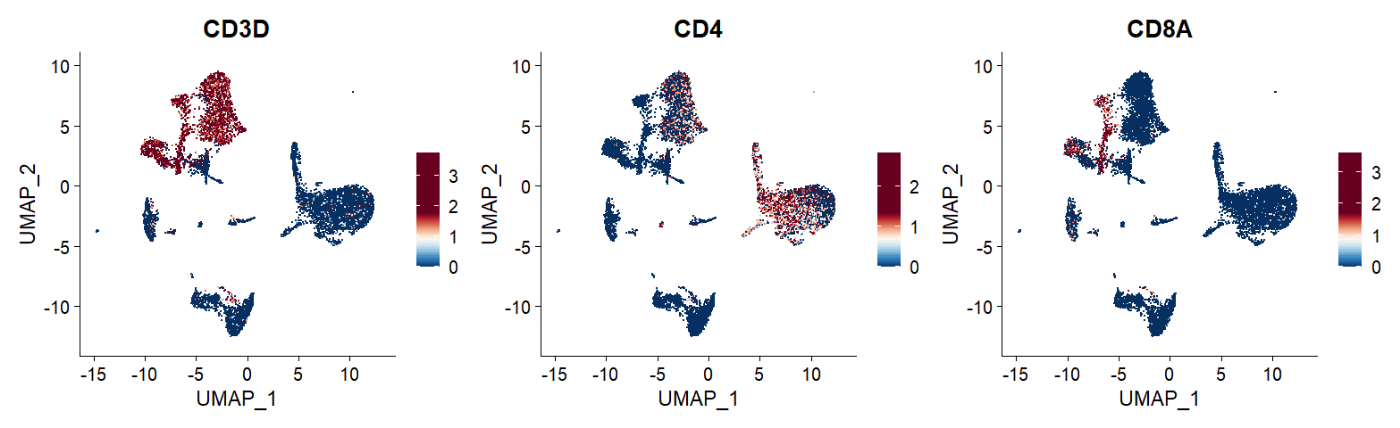
FeaturePlot(sc, features = c("CD3D","CD4","CD8A"), ncol = 3) &
scale_colour_gradientn(colours = rev(brewer.pal(n = 11, name = "RdBu")))

CD4やCD8の陽性細胞がぼんやりしている
対策1)order引数をTRUE
FeaturePlot()のorder=引数をTRUEにすると、陽性細胞が前面に表示される。
FeaturePlot(sc, features = c("CD3D","CD4","CD8A"), ncol = 3, order =T) &
scale_colour_gradientn(colours = rev(brewer.pal(n = 11, name = "RdBu")))

対策2)色を割り当てる範囲をlimitsで狭める
scale_color_gradientn()で色を修飾する際にlimits=引数で色を割り当てる範囲を指定する。
上限より大きい値を持つ細胞には色が割り当てられないので灰色に表示されてしまうが。
FeaturePlot(sc, features = c("CD3D","CD4","CD8A"), ncol = 3) &
scale_colour_gradientn(colours = rev(brewer.pal(n = 11, name = "RdBu")),
limits = c(0,1))

対策3)与える色ベクトルの最後の色を繰り返して、ある値以上は同じ色を持つようにする
色ベクトルの最後の要素を繰り返すように工夫する。例えば青 -> 黄 -> 赤 -> 赤 -> 赤 ,,,のようにしてやる。
col <- rev(brewer.pal(n = 11, name = "RdBu"))
col <- c(col, rep(col[length(col)], times = 11)) # timesの回数は好きに決定
FeaturePlot(sc, features = c("CD3D","CD4","CD8A"), ncol = 3) &
scale_colour_gradientn(colours = col)
DoHeatmapの値の範囲
plotに使用するslotを変更
DoHeatmap()ではデフォルトでscale.dataスロットの値をplotする。
slot="data"を指定すれば、FeaturePlot()やDotPlot()と同様の尺度で表示されるが、色合いの悪さが目立つようになる。
DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14"),
slot = "data")

slot="counts"でread count値をplotすることも可能。
DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14"),
slot = "counts")

表示域を変更 disp.max
カウント値はもっと大きな値を持っているはずだが、disp.max=引数がデフォルトで6となっているので、それ以上は頭打ちとなっている。scale.dataの場合はdisp.max=2.5が設定される。
disp.max=を調整すれば、dataスロットのデータでも見やすくできる。
DoHeatmap(sc, features = c("CD3D", "MS4A1", "CD14"),
slot = "data",
disp.max = 3)

※ ただし、disp.max=の調整では、指定値以上の発現値を持つ場合でも、発現量=指定値として表示されてしまうので誤った情報である。上の例だと、発現値上限3だと、黄色く表示された細胞の発現値が4なのか5なのかもっと多いのかの情報も無くなってしまう。
与える色ベクトルの最後の色を繰り返して、ある値以上は同じ色を持つようにする
この方法であれば、発現値がdisp.max=の値で頭打ちにならずに、嘘ではない情報を表現できる。
col <- viridis(n = 10)
col <- c(col, rep(col[length(col)], times = 5))
DoHeatmap(sc,
features = c("CD3D", "MS4A1", "CD14"),
slot = "data") +
scale_fill_gradientn(colours = col)

【表示目盛り】
ggplotの修飾で軸の目盛りを調整する例を紹介する。
デフォルトの目盛り幅だと、フィルタリングの際の閾値を決めにくいことがある。以下のFeatureScatter()の例だと、nCount_RNAの0の次の目盛りが20000となってしまっている。
FeatureScatter(sc,
feature1 = "nCount_RNA",
feature2 = "Percent_mito",
group.by = "orig.ident")

ggplotのscale_x_continuous()のbreaks=引数で表示目盛りを細かくすると良い。これなら2000-3000ぐらいを下限の閾値に設定すればいいことがわかる。
FeatureScatter(sc, feature1 = "nCount_RNA", feature2 = "Percent_mito", group.by = "orig.ident") +
scale_x_continuous(breaks = seq(0,max(sc$nCount_RNA), by=1000)) +
scale_y_continuous(breaks = seq(0,max(sc$Percent_mito), length.out = 20)) +
theme(axis.text.x = element_text(size = 6, angle = 90, hjust = 1),
axis.text.y = element_text(size = 6))

【Legend】
Seurat plotのlegendの調整例を紹介する。
凡例の位置
Seuratのplotの凡例は通常右側に表示されるが、ggplot2のtheme(legend.position=)で表示位置が変更できる。
legendの表示数がplot表示範囲から見切れる場合は、legendの文字サイズを変更するもの手だが、段組みを指定することでも対応できる。guides(color = guide_legend(nrow = 3))のようにすると、legendが3行で表示される。(※ color=なのはdotplotだから。VlnPlot()ではfill=になる。)
DimPlot(pbmc, label = TRUE, repel = TRUE) +
theme(legend.text = element_text(size = 12), # 凡例の文字サイズ
legend.position = "bottom" # 凡例の位置
) +
guides(color = guide_legend(nrow = 3)) # 凡例の行数、列数を指定

legend.position=でlegendの位置をマニュアルで指示するとplot枠内に配置することも可能。
DimPlot(pbmc, label = TRUE, repel = TRUE) +
theme(legend.text = element_text(size = 12),
legend.position = c(0.05,0.9), # legend位置を指定。c(x座標, y座標)
# legendに枠をつける。
legend.background = element_rect(fill = "lightyellow",
color = "black",
linewidth = 0.5)
) +
guides(color = guide_legend(nrow = 5, # 行数
label.vjust = 0.5 # textの位置調整
)
)

凡例内のドットサイズ
legend内のドットサイズがplot中のドットサイズのままで見えづらいことがある。plotのドットサイズとlegendのドットサイズを分けて指定するためには、override.aes=を使用する。
指定したいドットのパラメーターをlistで指定する。
DimPlot(pbmc, label = TRUE, repel = TRUE) +
theme(legend.text = element_text(size = 12),
legend.position = "bottom") +
guides(color = guide_legend(override.aes = list(size = 5, alpha = 0.5),
nrow = 3))

【ラスタライズ】
plotを論文や発表に使う用にローカルに書き出すときに、pdfやsvg拡張子で保存しておくと、adobe illustrator等で編集可能なデータを保存できる。Rのコマンド操作では修正しきれない部分やコマンドが冗長になる場合には手作業で修正した方が早い場合もある。
pdf("dimplot.pdf")
DimPlot(sc)
dev.off()
pdf("dimplot.svg")
DimPlot(sc)
dev.off()
adobe illustratorで開くと、DimPlot()の点ひとつひとつが編集可能なデータとなっている。

svg画像をillustratorで開いた画面
しかし、細胞数が多くなると如実にデータ処理が遅くなる。
Seuratのplotコマンドにはraster=引数が用意されており、raster=TRUEとするとplotが編集不可の焼き付いた画像が得られる。
pdf("dimplot.pdf")
DimPlot(sc, raster=TRUE)
dev.off()
pdf("dimplot.svg")
DimPlot(sc, raster=TRUE)
dev.off()
この画像は、軸やラベル等は編集可能でplot領域のデータのみが焼き付いたものとなっている。

svg画像をillustratorで開いた画面。dotは焼き付くが、軸やラベルは編集可能
デフォルトでは元のplotが非常に粗くなってしまう。必要に応じてraster.dpi=引数から解像度を変更する。デフォルトではraster.dpi=c(512,512)である。単位がわかりにくいが、dpiなので縦512ピクセル/インチ、横512ピクセル/インチということだろう。

raster.dpi = c(256, 256)

raster.dpi = c(1024, 1024)
raster.dpi=を上げても点がガタガタなままの場合は、pt.size=で点の大きさを上げてやると良い感じになる。
pdf("dimplot3-2.pdf")
DimPlot(sc, raster = TRUE, raster.dpi = c(1024, 1024), pt.size = 2)
dev.off()

以降はSeurat plotの種類ごとにtipsを紹介する。
【VlnPlot】
細胞数が多い時のVlnPlotはpt.size = 0を指定
細胞数が多いデータの場合、点が重なりすぎてviolin plotが見づらくなる。
VlnPlot(sc,
features = c("nFeature_RNA", "nCount_RNA", "Percent_mito"),
group.by = "orig.ident")

VlnPlot(sc,
features = c("nFeature_RNA", "nCount_RNA", "Percent_mito"),
pt.size = 0,
group.by = "orig.ident")

見たい遺伝子数が多い時はstack引数が有用
VlnPlot(sc,
features = c("CD3D","CD4","CD8A","PTPRC","MS4A1","CD19","CD79A","CD14","CD68","ITGAX"),
stack = T)

さらにflip=引数もTRUEにした方が見栄えが良い。
VlnPlot(sc,
features = c("CD3D","CD4","CD8A","PTPRC","MS4A1","CD19","CD79A","CD14","CD68","ITGAX"),
stack = T,
flip = T)

【DimPlot】
特定の細胞をハイライト表示する cells.highlight
特定の細胞のみを色付きでplotすると、どこに分布しているのかを確認しやすい。
DimPlot()のcells.highlight=引数に細胞バーコードのベクトルを指定する。
DimPlot(sc, cells.highlight = c("AAATGGATCGTAGGGA-1"))

WhichCells
特定の条件を満たす細胞バーコードベクトルを取得するにはWhichCells()機能が有用である。
DimPlot(sc,
cells.highlight = WhichCells(sc, idents = c("1","3")))

WhichCells()のidents=引数は、active.identスロットに保存された名前を対象にする。
active.identの内容はlevels()関数などで確認可能である。
levels(sc)
[1] "0" "1" "2" "3" "4" "5" "6" "7" "8" "9"
WhichCells()の指定方法いろいろ
WhichCells()のexpression=引数を使えば、active.identを変えずにmeta.dataスロットを参照できる。
expression = meta.data列名 %in% 欲しいlevel と書く。meta.data列名は""が要らないのと、levelが複数ある場合は==ではなく%in%とする必要がある。
Idents(sc) <- "cluster0.5"
DimPlot(sc,
cells.highlight = WhichCells(sc, expression = cluster0.1 %in% c("1","3")))

またexpression=引数を使えば、遺伝子発現量の閾値で細胞バーコードを取り出すことができる。
DimPlot(sc,
cells.highlight = WhichCells(sc, expression = CD8A>0)
)

DimPlot(sc,
cells.highlight = WhichCells(sc, expression = CD8A>0 & GZMA>1)
)

複数ハイライトを色分けして表示
WhichCells()で複数条件を満たす細胞バーコードを取り出しても、1つのハイライトになってしまう。
各条件で複数ハイライトを作りたければcells.highlight=引数に細胞バーコードのリストを指定する。
listの要素に名前をつけていなければ、legendがGroup_1, Group_2,,,になる。
cols.highlight=引数でlist数に対応した色ベクトルを用意しなければ全て同じハイライト色になるので注意。
DimPlot(sc,
cells.highlight = list(cluster1 = WhichCells(sc, expression = cluster0.1 %in% "1"),
cluster3 = WhichCells(sc, expression = cluster0.1 %in% "3")),
cols.highlight = c("darkred", "darkblue")
)

Dotに枠線を付ける
ggplotのdotの形状番号21-25は枠線を持つ。DimPlot()にはdot shapeを指定する引数が用意されていないので、次のようにplotに使用する値(ここではumapの値)と色分けに使用する情報(cluster名やcluster番号)などを取り出して、ggplotで書き直すことで簡単に作図できる。
枠がある形状の場合、色分けに列はcol=ではなくfill=に指定する。col=で指定した場合、枠線の色が群によって分けられる。
df <- data.frame(sc@reductions[["umap"]]@cell.embeddings,
cluster = as.factor(sc@active.ident))
# geom_pointのshape引数に塗りつぶし可能なshape番号(21~25)を指定。
ggplot(df, aes(x = umap_1, y = umap_2, fill = cluster)) +
geom_point(shape = 21) +
theme_classic()

【DoHeatmap】
上部ラベルが見切れる対策
ヒートマップ上部にラベルされるクラスター名が長いと、文字が途中で見切れてしまう。
DEGs <- FindAllMarkers(pbmc, only.pos = T, logfc.threshold = 1)
DEGs %>%
group_by(cluster) %>%
top_n(n = 5, wt = avg_log2FC) -> top5
DoHeatmap(pbmc, features = top5$gene)

1. フォントサイズの変更
size=引数で上部ラベルのフォントサイズのみを指定できる。
ただ、ラベルの文字数が多い時はフォントサイズをかなり小さくしても見切れてしまう。
DoHeatmap(pbmc, features = top5$gene, size = 2)

凡例でどの列がどの細胞かはわかるので、ラベルが不要だというときは、label=FALSEにするとよい。
DoHeatmap(pbmc, features = top5$gene, label = F)

2. plot marginを調整
ggplot2のplot margin=調節を使用すると改善可能。上、右、下、左の順で余白を指定する。
DoHeatmap(pbmc, features = top5$gene, size = 4) +
theme(plot.margin = unit(c(2,0,0,0),"cm"))

Discussion