【RNAseq】[immunedeconv] RNAseqデータから免疫細胞比率を推定
はじめに
免疫細胞など発現プロファイルが良く知られた細胞では、bulk RNAseqデータからその存在比率や存在量を推定することができる。
有名なのはCIBERSORTであるが、そのほかにもTIMER、MCPCounter、xCell、EPICなどがあり、それぞれ異なる手法で異なる細胞種を推定している。
これらはまだシングルセル解析が一般的になる前の2015-2017年ごろに報告されている。
各ツールごとにRパッケージが公開されているが、本記事では各推定ツールを1つのRパッケージにまとめた「immunedeconv」パッケージを紹介する。
参考
https://omnideconv.org/immunedeconv/articles/immunedeconv.html
https://academic.oup.com/bioinformatics/article/35/14/i436/5529146?login=false
アルゴリズム
https://omnideconv.org/immunedeconv/articles/immunedeconv.html
手法は大きく2つのカテゴリに分かれる
-
細胞マーカー遺伝子のエンリッチメントスコア
ある細胞種の存在量が多ければ、bulk RNAseqデータでもその細胞マーカーの発現は高いはずである。これをスコア化して存在量の指標とする。 -
Deconvolution
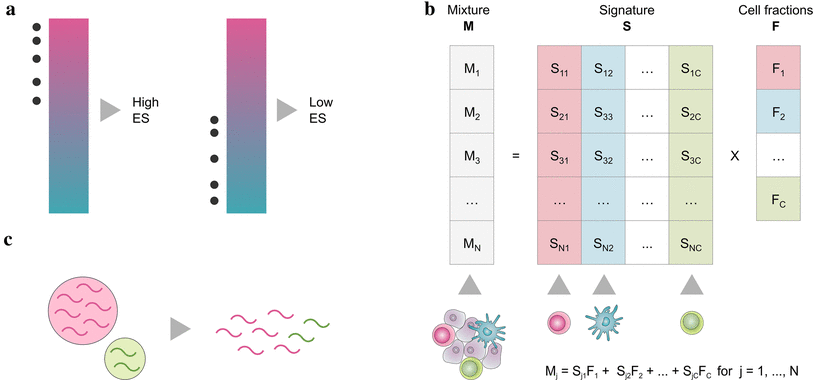
直訳すると逆畳み込みで意味がわかりにくいが、上図bを見ると想像しやすい。
bulk RNAseqデータ(M)が細胞種シグネチャ(S)とその細胞存在比率(F)の行列積で求められるとする。この式から、bulk RNAseqデータと細胞種シグネチャがあれば、細胞存在比率が導出できるのがわかる。この導出をdeconvolutionと称している。
現在ではシングルセル解析データを使えば細胞特異的な遺伝子シグネチャを簡単に取得できるが、今でも上記ツールは現役で使用できる。
入力データ
行に遺伝子、列にサンプルを配置した発現マトリクスを使用する。遺伝子はHGNC gene symbolで表記する。
遺伝子IDなどから変換する方法はこちらの記事で紹介している。
入力に用いるbulk RNAseqデータは、TPM正規化したものを想定している。TPM正規化後にさらなる対数変換は推奨されていない。
ただし、xCellは発現ランキング、MCP-counterはマーカー遺伝子の平均発現量を使用するため発現量正規化方法はさほど重要ではない。
インストール
まずはパッケージをインストールする。Githubページで公開されており、インストールにはremotesパッケージがまずは必要となる。
install.packages("remotes")
remotes::install_github("icbi-lab/immunedeconv")
library(immunedeconv)
初回インストール時には、xCell/EPIC/MCPcounter/mMCPcounter/ConsensusTME,,,など内部で使用する推定ツールもインストールされる。
都度、関連パッケージの更新について聞いてくるので、適当に答えて進める。
使用可能なツール
deconvolution_methodsというオブジェクトにツール一覧が保存されている。
2023年8月現在、以下のツールが指定できる。(この1年でabis/consensus_tme/estimateが追加されている。)

使用可能な推定ツール
マウスのデータの場合は以下の4つが使用できる。
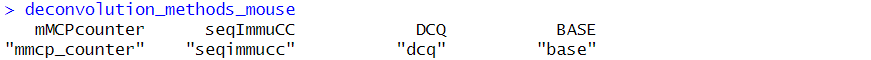
使用可能な推定ツール マウス
CIBERSORTのソースコード取得
CIBERSORTのソースコード取得
まずはRegisterからユーザー登録を行う。※ アカデミア向けに公開されている。
数年前まで、CIBERSORTとCIBERSORTxのそれぞれのウェブページが閲覧できたが、2023年8月現在ではCIBERSORTのページは見れなくなり、CIBERSORTxのページのみになっている。
CIBERSORTのsource codeはアーカイブ化されてCIBERSORTxのページからアクセスできるようだ。Menu > CS Archive > CS Download
CIBERSORT Archiveページを下にスクロールしていき、Request CIBERSORT source codeのリンクをクリック。

CIBERSORT Archiveページ
Yesで進めると、数日以内にメールで届くという旨のメッセージが表示される。待つしかない。

届いたメールのリンクからソースコードをダウンロードする。

届いたメール
22種類の免疫細胞シグネチャのLM22.txtも同ページからダウンロードしておく。
デモデータ
immunedeconv提供のデモデータを使用する。このデモデータには4例のbulkRNAseqデータが含まれており、フローサイトメーターでB細胞/NK細胞/CD4 T細胞/CD8 T細胞の比率を計測した正解となるデータもセットになっている。
# デモデータの呼び出し
demo <- dataset_racle
# 正解データ
frac <- demo$ref
library(ggplot2)
ggplot(data = frac, aes(x = sample, y = true_fraction, fill = cell_type)) +
geom_col()

bulkRNAseqデータは32467遺伝子 x 4検体の発現マトリクスとして提供されていた。
# 発現マトリクス
mat <- demo$expr_mat
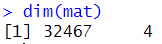
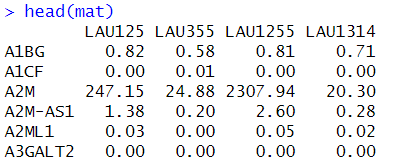
データ分布を見ると、各検体の合計発現量が100万ちかいので、TPM変換した後に遺伝子フィルタリングをしたのだろう。追加でTPM変換処理などは行わずに進めてみる。

推定の実行
deconvolution()機能を使用する。gene_expression=引数に発現マトリクスを、method =引数に推定ツールを指定する。
推定ツール指定時の文字列は上述のdeconvolution_methodsから確認できる。
さらなるオプション類は使用する推定ツールによって変わる。
deconvolution_ツール名()という機能も用意されている。こちらからの方が、ツール専用のオプションを確認しやすい。
※gene_expression=引数ではなく、gene_expression_matrix=引数に発現マトリクスを指定する。

MCPcounter
発現マトリクスと遺伝子表記を指定する。デフォルトはfeature_types = "HUGO_symbols"。"affy133P2_probesets"と"ENTREZ_ID"も選択できる。
res_MCPcounter <- deconvolute_mcp_counter(
gene_expression_matrix = mat,
feature_types = "HUGO_symbols"
)
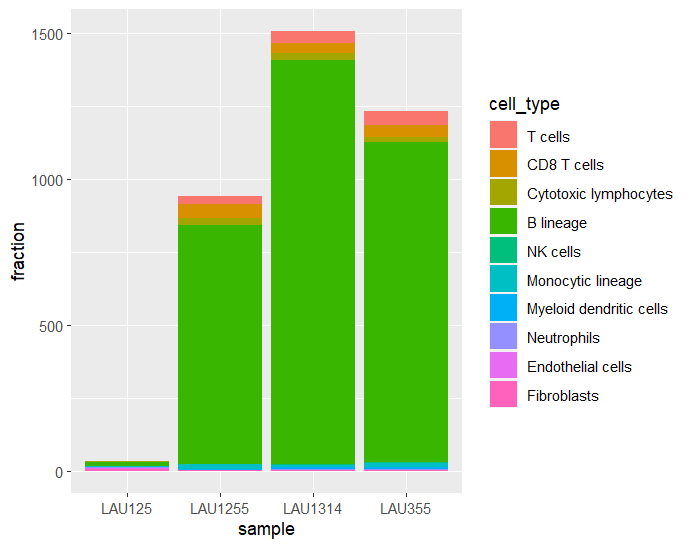
10種類の細胞が推定される。結果は細胞の比率ではなく、細胞スコアとなる。

df <- reshape2::melt(res_MCPcounter)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col() +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355"))
EPIC
EPICにはTumor用とそれ以外用のシグネチャが用意されている。tumor=引数からTRUE/FALSEで変更する。scale_mrna=はTRUEが推奨とのこと。
内部で使用するEPIC()機能のhelp
https://rdrr.io/github/GfellerLab/EPIC/man/EPIC.html
以下の引数で、deconvolute(gene_expression = mat, method = "epic")と同じ推定結果が得られる。(つまりこれがデフォルト。Tumor用がTRUEになっている。)
res_EPIC <- deconvolute_epic(
gene_expression_matrix = mat,
tumor = TRUE,
scale_mrna = TRUE
)
8種類の細胞の存在比率が予測される。

df <- reshape2::melt(res_EPIC)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col() +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355"))
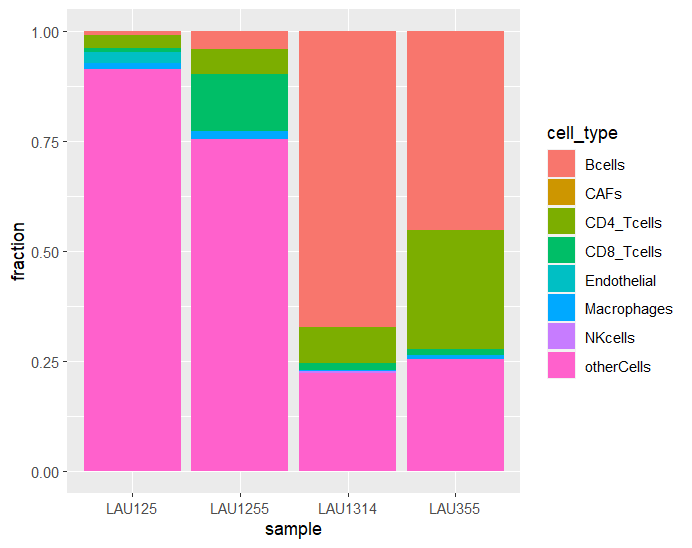
tumor=TRUE
ちなみにtumor=FALSEとすると次のような結果が得られた。CAFが予測から無くなっている。
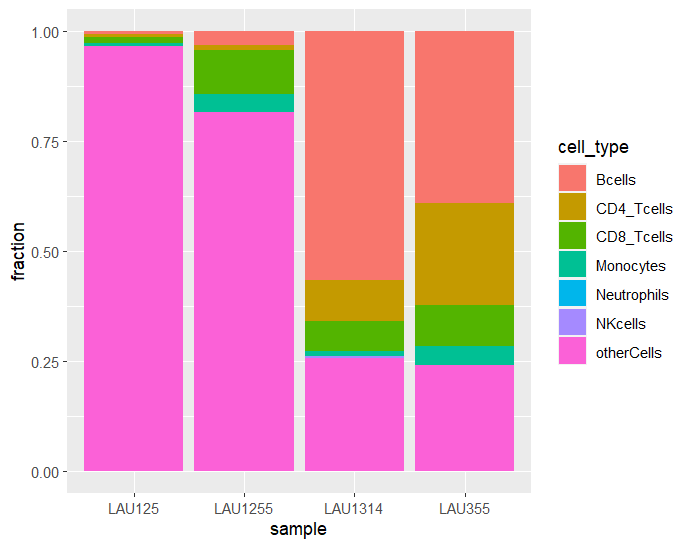
tumor=FALSE
quanTIseq
こちらもEPICと同様でtumor=引数とscale_mrna=がある。
内部で使うrun_quantiseq()機能のhelp
https://rdrr.io/github/federicomarini/quantiseqr/man/run_quantiseq.html
次の引数でdeconvolute(gene_expression = mat, method = "quantiseq")と同じ推定結果が得られる。
res_quanTIseq <- deconvolute_quantiseq(
gene_expression_matrix = mat,
tumor = TRUE,
arrays = FALSE, # RNAseqではなくmicroarrayの場合はTRUE
scale_mrna = TRUE
)
11種類の細胞の存在比率が予測される。

df <- reshape2::melt(res_quanTIseq)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col() +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355"))
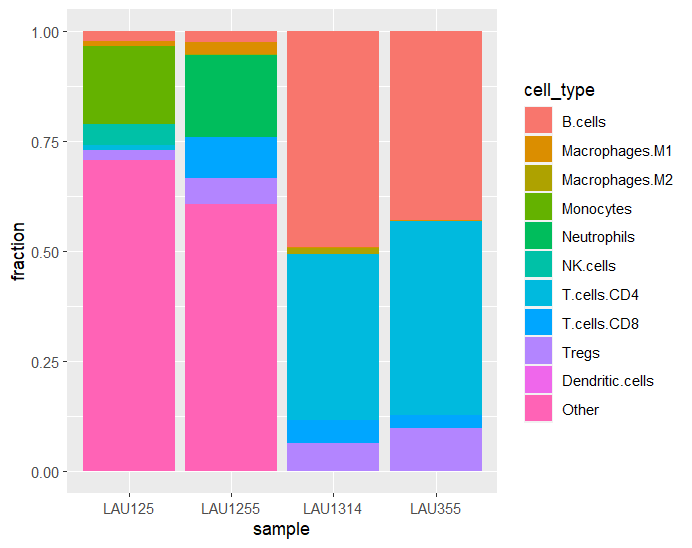
tumor=TRUE
ちなみにtumor=FALSEとすると次のような結果が得られた。今回のデモデータでは大きく変わることは無かった。

tumor=FALSE
xCell
ssGSEAを使って細胞マーカーの発現ランク値を元にスコアリングする。ssGSEAについてはこちらの記事で紹介した。
内部で使うxCellAnalysis()機能のhelp
https://rdrr.io/github/dviraran/xCell/man/xCellAnalysis.html
expected_cell_types=NULLで推測できる全ての細胞を計算する。
res_xCell <- deconvolute_xcell(
gene_expression_matrix = mat,
arrays = FALSE, # RNAseqではなくmicroarrayの場合はTRUE
expected_cell_types = NULL
)
67種類の細胞がスコアリングされる。
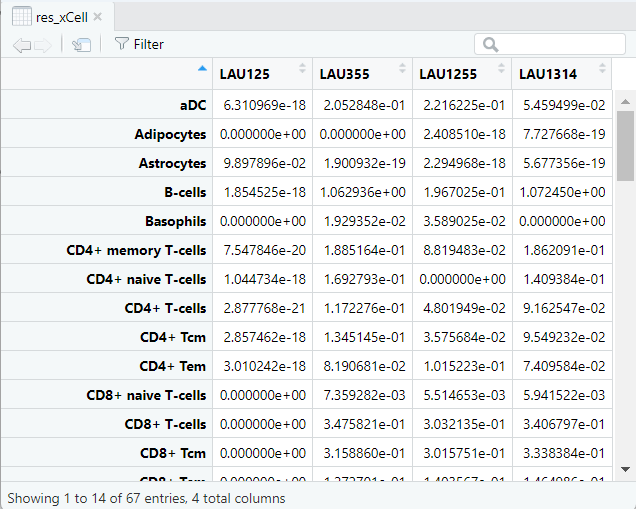
pheatmap::pheatmap(t(res_xCell))

自身のbulk RNAseqデータに含まれないような細胞を除くためにexpected_cell_types=で対象の細胞を指定すればよいが、指定の仕方がよくわからなかった。。。
CIBERSORT
公式サイトから承認を得てダウンロードしたソースコードと細胞シグネチャのファイルパスを事前に設定する。
set_cibersort_binary("/path/to/CIBERSORT.R")
set_cibersort_mat("/path/to/LM22.txt")
CIBERSORTには細胞比率(合計が1)と細胞存在量を推定する2つのモードがある。absolute=引数のTRUE/FALSEで切り替える。
abusolute=FALSE
res_CIBERSORT <- deconvolute_cibersort(
gene_expression_matrix = mat,
arrays = FALSE,
absolute = FALSE
)
22種類の免疫細胞比率が推測される。

df <- reshape2::melt(res_CIBERSORT)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col(color = "black") +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355")) +
guides(fill = guide_legend(ncol = 1))
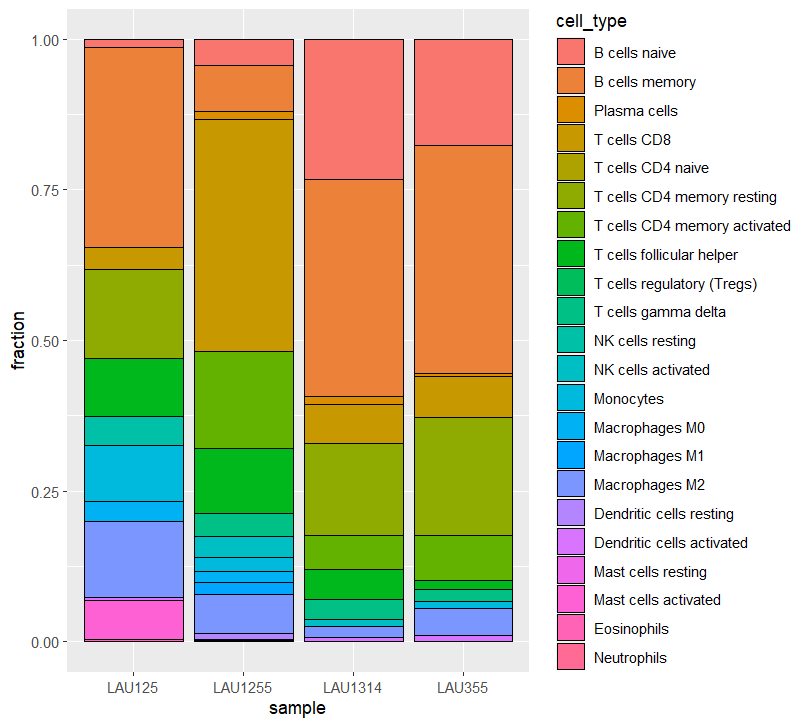
absolute = FALSE
abusolute=TRUE
res_CIBERSORTx <- deconvolute_cibersort(
gene_expression_matrix = mat,
arrays = FALSE,
absolute = TRUE
)
df <- reshape2::melt(res_CIBERSORTx)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col(color = "black") +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355")) +
guides(fill = guide_legend(ncol = 1))
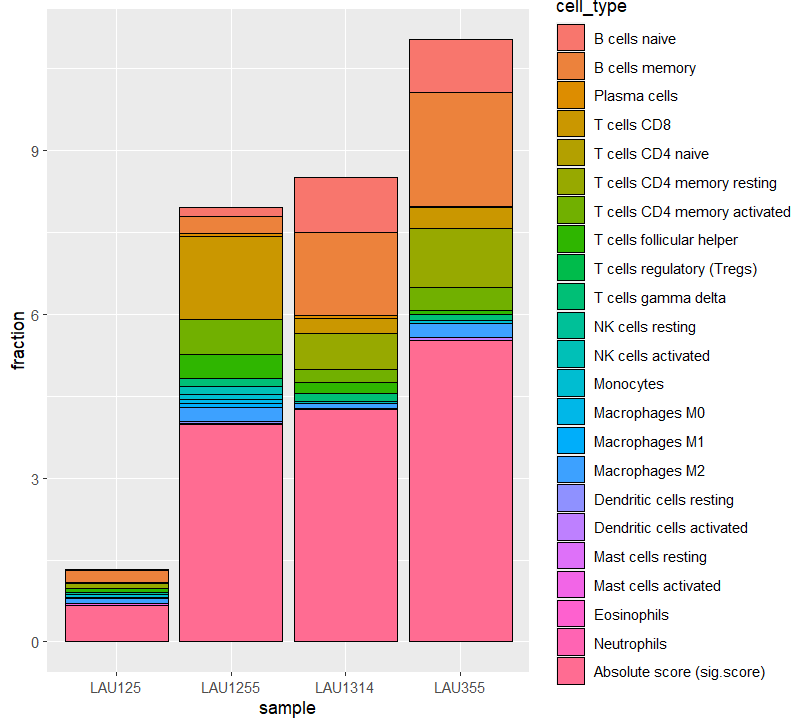
absolute = TRUE
TIMER
TIMERではindications=引数で腫瘍の種類を指定しなければならない。指定名は以下を確認。

TCGAでの腫瘍名の省略形となっている。次のリンクから確認できる。
https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations
指定する際は、検体の数だけ指定する。このデモデータはmelanomaのものとのことだったので、SKCM (Skin Cutaneous Melanoma)を使用してみた。
res_TIMER <- deconvolute_timer(
gene_expression_matrix = mat,
indications = rep("skcm", ncol(mat))
)
6種類の細胞の値が推定された。合計すると1に近いように見えるが、スコアである。

df <- reshape2::melt(res_TIMER)
colnames(df) <- c("cell_type", "sample", "fraction")
ggplot(data = df, aes(x = sample, y = fraction, fill = cell_type)) +
geom_col() +
scale_x_discrete(limits = c("LAU125", "LAU1255", "LAU1314", "LAU355"))
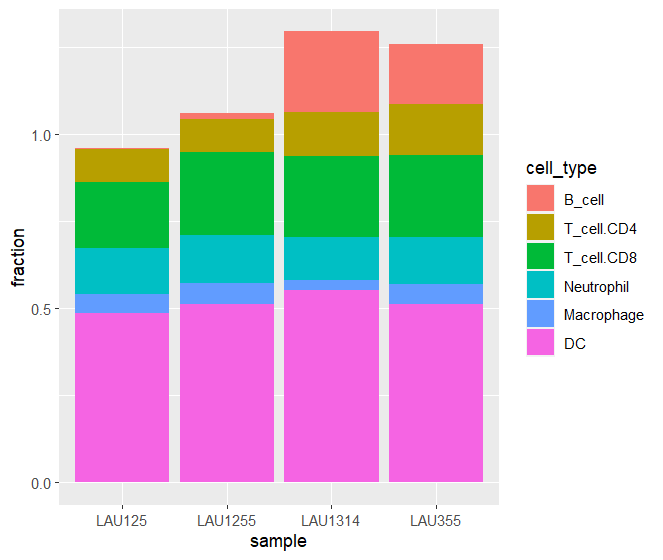
ConsensusTME
腫瘍微細環境のスコアリングツール。
こちらもTIMER同様indications=引数で腫瘍の種類を指定しなければならない。
計算方法はmethod=引数から変更可能。デフォルトが"ssgsea"。その他にも "gsva", "plage", "zscore", "singScore"が選択可能。"singScore"以外はGSVAパッケージの引数となっている。
method="singScore"の場合はsingscoreパッケージのmultiScore()機能が使用される。
https://davislaboratory.github.io/singscore/reference/multiScore.html
res_ConsensusTME <- deconvolute_consensus_tme(
gene_expression_matrix = mat,
indications = rep("skcm", ncol(mat)),
method = "ssgsea"
)
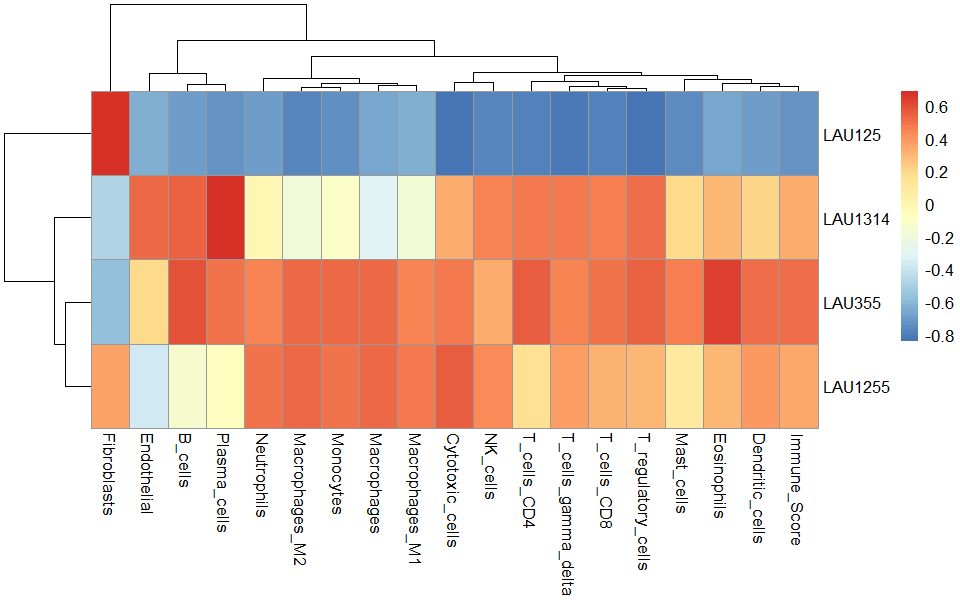
19種類の細胞がスコアリングされる。ssGSEAやGSVAの場合はマイナスの値を取ることもある。
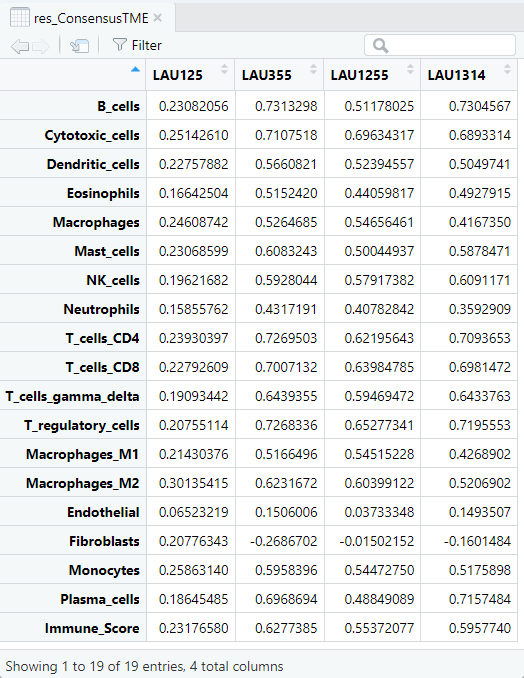
pheatmap::pheatmap(t(res_ConsensusTME))
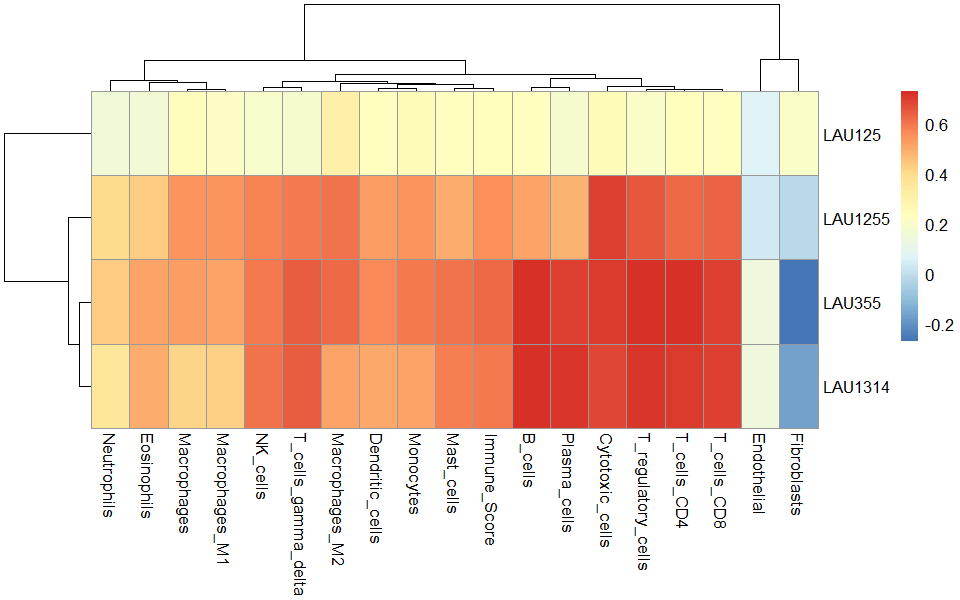
method="ssgsea"
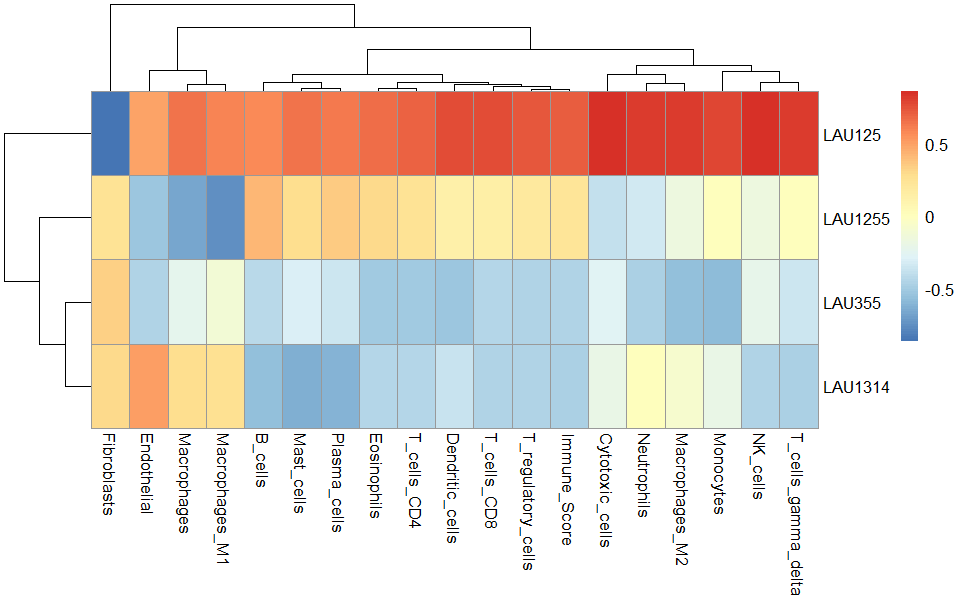
method="gsva"
method="plage"
method="zscore"
method="singScore"を指定すると、なぜか以下のエラーが出た。
Error in
colnames<-(*tmp*, value = colnames(gene_expression_matrix)) :
次元が 2 未満のオブジェクトに 'colnames' を設定しようとしました
しかし、内部で動いているConsensusTMEパッケージのconsensusTMEAnalysis()機能を使えばワークした。cancerType=引数はサンプルの数だけ指定するのではなく、1つの腫瘍タイプを大文字で指定する。
res_ConsensusTME <- ConsensusTME::consensusTMEAnalysis(
bulkExp = mat,
cancerType = "SKCM",
statMethod = "singScore"
)
pheatmap::pheatmap(t(res_ConsensusTME$Scores))
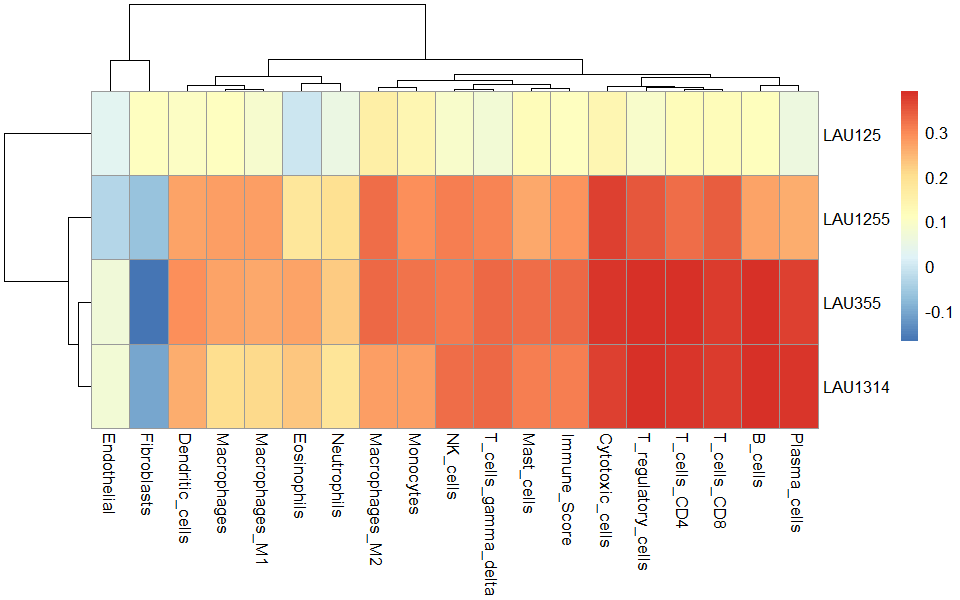
method="singScore"
ABIS
あまり見ないが、ABsolute Immune Signal (ABIS) deconvolutionというツール
res_ABIS <- deconvolute_abis(
gene_expression_matrix = mat,
arrays = FALSE # microarrayの場合はTRUE
)
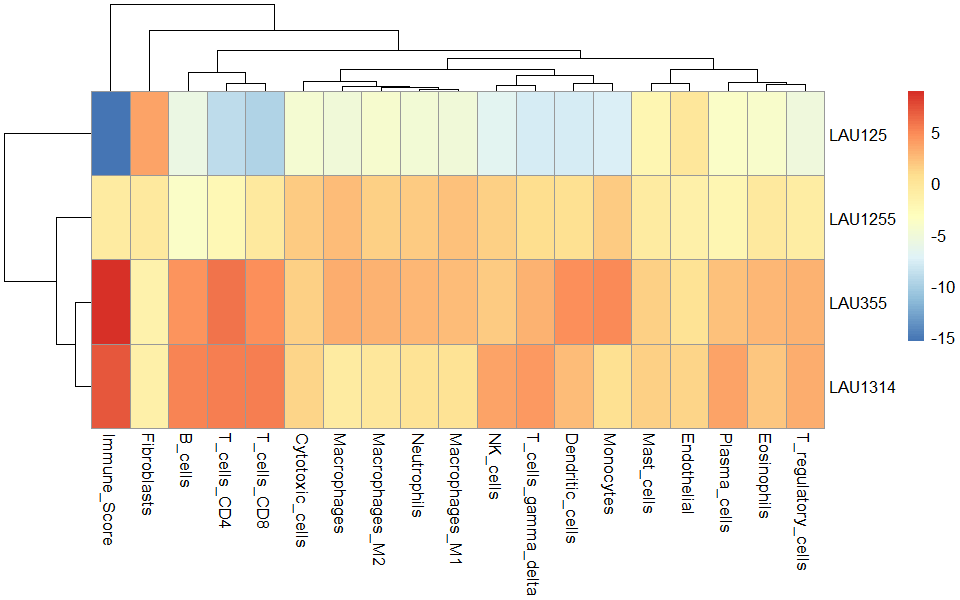
17種類の細胞のスコアリングがされる。スコアはマイナスの値も取る。

pheatmap::pheatmap(t(res_ABIS))

Githubページには0-100のパーセントを計算しているとある。マイナスの値は0とするとか。

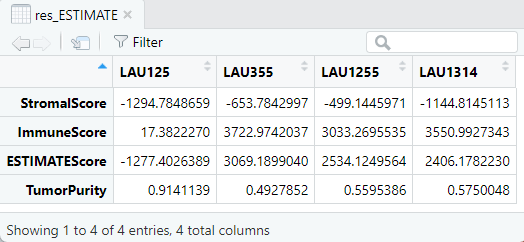
ESTIMATE
ESTIMATEはRNAseqデータから腫瘍純度をスコアリングするツールで、上記までとは少し毛色が異なる。
オプションとなる引数は無い。
res_ESTIMATE <- deconvolute_estimate(
gene_expression_matrix = mat
)
結果は、間質スコアと免疫細胞スコアから算出したESTIMATE scoreを元に腫瘍純度も出してくれる。
値の解釈
様々な推定ツールが異なった値を返しているが、どれが正しい正しくないは断定できない。どうしてもスコアや存在比が、他の検体と比べて高いか低いか、他の細胞種と比べて高いか低いかという言及が必要になることが多い。
その場合に、上記ツールの値は細胞種間で比較してよいものか、検体間で比較してよいものかは知っておくべきだ。
immunedeconvの公式ページにヒントがある。

https://omnideconv.org/immunedeconv/articles/immunedeconv.html
Discussion