【R】 遺伝子ID/遺伝子名の変換方法
遺伝子の表記には、遺伝子名だけでなく各データベースごとの遺伝子IDがある。遺伝子名であっても複数存在したり、逆に遺伝子IDがあっても遺伝子名が付いていないものもある。
わかりやすく遺伝子名で解析を進めたいところであるが、解析パッケージによって求められる遺伝子表記が異なり、その度に 「遺伝子名 ⇄ 遺伝子ID」 変換 や 「あるデータベースの遺伝子ID ⇄ 違うデータベースの遺伝子ID」 変換が求められる。
本記事では遺伝子表記の変換方法を幾つか紹介する。
各生物種の遺伝子アノテーション情報をまとめたデータベースがRのパッケージとして提供されている。ここから遺伝子アノテーションに加え、パスウェイデータベースなどのパッケージも探すことができる。
【 Genome wide annotationパッケージ 】
上記リンクからorg.生物種.eg.dbというパターンのパッケージを見つけてインストールしておく。ヒトの場合はorg.Hs.eg.db、マウスの場合はorg.Mm.eg.dbを使用する。
(egはEntrez geneの略??)
library(org.Hs.eg.db)
一度パッケージを読み込むと、パッケージ名のオブジェクトが参照できるようになる。
試しにオブジェクト名を打ってみると、データベース内の各情報ソースの名前やversionなどが確認できる。
org.Hs.eg.db
データベースオブジェクトの中身
OrgDb object:
| DBSCHEMAVERSION: 2.1
| Db type: OrgDb
| Supporting package: AnnotationDbi
| DBSCHEMA: HUMAN_DB
| ORGANISM: Homo sapiens
| SPECIES: Human
| EGSOURCEDATE: 2022-Mar17
| EGSOURCENAME: Entrez Gene
| EGSOURCEURL: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA
| CENTRALID: EG
| TAXID: 9606
| GOSOURCENAME: Gene Ontology
| GOSOURCEURL: http://current.geneontology.org/ontology/go-basic.obo
| GOSOURCEDATE: 2022-03-10
| GOEGSOURCEDATE: 2022-Mar17
| GOEGSOURCENAME: Entrez Gene
| GOEGSOURCEURL: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA
| KEGGSOURCENAME: KEGG GENOME
| KEGGSOURCEURL: ftp://ftp.genome.jp/pub/kegg/genomes
| KEGGSOURCEDATE: 2011-Mar15
| GPSOURCENAME: UCSC Genome Bioinformatics (Homo sapiens)
| GPSOURCEURL:
| GPSOURCEDATE: 2022-Nov23
| ENSOURCEDATE: 2021-Dec21
| ENSOURCENAME: Ensembl
| ENSOURCEURL: ftp://ftp.ensembl.org/pub/current_fasta
| UPSOURCENAME: Uniprot
| UPSOURCEURL: http://www.UniProt.org/
| UPSOURCEDATE: Fri Apr 1 14:42:16 2022
アノテーションパッケージを読み込むと、合わせてAnnotationDbiパッケージが読み込まれる。
データベースからのデータ取り出しにはAnnotationDbiで用意された関数を使用することになる。
項目確認
データベースに存在する項目名を確認するにはkeytypes()機能かcolumns()機能を使用する。
keytypes(org.Hs.eg.db)
[1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "ENTREZID" "ENZYME"
[8] "EVIDENCE" "EVIDENCEALL" "GENENAME" "GENETYPE" "GO" "GOALL" "IPI"
[15] "MAP" "OMIM" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" "PMID"
[22] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" "UNIPROT"
ENSEMBLデータベースでの遺伝子ID, EntreZ ID, 遺伝子名 (SYMBOL)だけでなく、Gene ontologyなどの情報もある。
各項目の説明はここにあった。
項目の説明
| Keytype | Description |
|---|---|
| ACCNUM | GenBank accession numbers |
| ALIAS | Commonly used gene symbols |
| ARACYC | KEGG Identifiers for arabidopsis as indicated by aracyc |
| ARACYCENZYME | Aracyc enzyme names as indicated by aracyc |
| CHR | Chromosome (deprecated for Bioc > 3.1) For this information you should look at a TxDb or OrganismDb object and search for an appropriate field like TXCHROM, EXONCHROM or CDSCHROM. This information can also be retrieved from these objects using an appropriate range based accesor like transcripts, transcriptsBy etc. |
| CHRLOC | Chromosome and starting base of associated gene (deprecated for Bioc > 3.1) For this information you should look at a TxDb or OrganismDb object and search for an appropriate field like TXSTART etc. or even better use the associated range based accessors like transcripts or transcriptsBy to get back GRanges objects. |
| CHRLOCEND | Chromosome and ending base of associated gene (deprecated for Bioc > 3.1) For this information you should look at a TxDb or OrganismDb object and search for an appropriate field like TXEND etc. or even better use the associated range based accessors like transcripts or transcriptsBy to get back GRanges objects. |
| COMMON | Common name |
| DESCRIPTION | The description of the associated gene |
| ENSEMBL | The ensembl ID as indicated by ensembl |
| ENSEMBLPROT | The ensembl protein ID as indicated by ensembl |
| ENSEMBLTRANS | The ensembl transcript ID as indicated by ensembl |
| ENTREZID | Entrez gene Identifiers |
| ENZYME | Enzyme Commission numbers |
| EVIDENCE | Evidence codes for GO associations with a gene of interest |
| EVIDENCEALL | Evidence codes for GO (includes less specific terms) |
| GENENAME | The full gene name |
| GO | GO Identifiers associated with a gene of interest |
| GOALL | GO Identifiers (includes less specific terms) |
| INTERPRO | InterPro identifiers |
| IPI | IPI accession numbers |
| MAP | cytoband locations |
| OMIM | Online Mendelian Inheritance in Man identifiers |
| ONTOLOGY | For GO Identifiers, which Gene Ontology (BP, CC, or MF) |
| ONTOLOGYALL | Which Gene Ontology (BP, CC, or MF), (includes less specific terms) |
| ORF | Yeast ORF Identifiers |
| PATH | KEGG Pathway Identifiers |
| PFAM | PFAM Identifiers |
| PMID | Pubmed Identifiers |
| PROBEID | Probe or manufacturer Identifiers for a chip package |
| PROSITE | Prosite Identifiers |
| REFSEQ | Refseq Identifiers |
| SGD | Saccharomyces Genome Database Identifiers |
| SMART | Smart Identifiers |
| SYMBOL | The official gene symbol |
| TAIR | TAIR Identifiers |
| UNIGENE | Unigene Identifiers |
| UNIPROT | Uniprot Identifiers |
データ取り出し
各項目の中身を確認するにはkeys()機能を使用する。
keys(データベースオブジェクト, keytype = "項目名")
試しにorg.Hs.eg.dbの全項目をチラ見してみる。
for(i in columns(org.Hs.eg.db)){
print(i)
str(keys(org.Hs.eg.db,i))
}
各項目の中身
[1] "ACCNUM"
chr [1:806658] "AA484435" "AAH35719" "AAL07469" "AB073611" "ACJ13639" "AF414429" "AI022193" "AK055885" ...
[1] "ALIAS"
chr [1:131663] "A1B" "ABG" "GAB" "HYST2477" "A1BG" "A2MD" "CPAMD5" "FWP007" "S863-7" "A2M" "A2MP" "A2MP1" ...
[1] "ENSEMBL"
chr [1:38636] "ENSG00000121410" "ENSG00000175899" "ENSG00000256069" "ENSG00000171428" "ENSG00000156006" ...
[1] "ENSEMBLPROT"
chr [1:21427] "ENSP00000286479" "ENSP00000428416" "ENSP00000483300" "ENSP00000482269" "ENSP00000478547" ...
[1] "ENSEMBLTRANS"
chr [1:38893] "ENST00000543404" "ENST00000566278" "ENST00000545343" "ENST00000544183" "ENST00000286479" ...
[1] "ENTREZID"
chr [1:66102] "1" "2" "3" "9" "10" "11" "12" "13" "14" "15" "16" "17" "18" "19" "20" "21" "22" "23" "24" "25" ...
[1] "ENZYME"
chr [1:975] "2.3.1.5" "2.3.1.87" "6.1.1.7" "2.6.1.22" "2.6.1.19" "2.7.10.2" "1.4.3.22" "2.4.1.37" "2.4.1.40" ...
[1] "EVIDENCE"
chr [1:20] "ND" "HDA" "IDA" "TAS" "IEA" "IBA" "IPI" "IMP" "NAS" "ISS" "IEP" "IGI" "IC" "EXP" "HEP" "HMP" "RCA" ...
[1] "EVIDENCEALL"
chr [1:20] "ND" "HDA" "IDA" "TAS" "IEA" "IBA" "IPI" "IMP" "NAS" "ISS" "IEP" "IC" "IGI" "EXP" "HMP" "HEP" "RCA" ...
[1] "GENENAME"
chr [1:65232] "alpha-1-B glycoprotein" "alpha-2-macroglobulin" "alpha-2-macroglobulin pseudogene 1" ...
[1] "GENETYPE"
chr [1:11] "protein-coding" "pseudo" "other" "unknown" "ncRNA" "tRNA" "rRNA" "scRNA" "snoRNA" "snRNA" ...
[1] "GO"
chr [1:18644] "GO:0003674" "GO:0005576" "GO:0005615" "GO:0008150" "GO:0031093" "GO:0034774" "GO:0062023" ...
[1] "GOALL"
chr [1:22834] "GO:0003674" "GO:0005575" "GO:0005576" "GO:0005615" "GO:0005622" "GO:0005737" "GO:0008150" ...
[1] "IPI"
chr [1:37945] "IPI00644018" "IPI00022895" "IPI00478003" "IPI00644361" "IPI00930394" "IPI00004421" ...
[1] "MAP"
chr [1:2003] "19q13.43" "12p13.31" "8p22" "14q32.13" "3q25.1" "2q35" "17q25.1" "16q22.1" "19q13" "19q13-qter" ...
[1] "OMIM"
chr [1:22739] "138670" "103950" "108345" "243400" "612182" "107280" "600338" "603488" "600950" "601065" ...
[1] "ONTOLOGY"
chr [1:3] "MF" "CC" "BP"
[1] "ONTOLOGYALL"
chr [1:3] "MF" "CC" "BP"
[1] "PATH"
chr [1:229] "04610" "00232" "00983" "01100" "00380" "00970" "00250" "00280" "00410" "00640" "00650" "02010" ...
[1] "PFAM"
chr [1:6280] "PF13895" "PF07678" "PF07677" "PF17791" "PF00207" "PF01835" "PF07703" "PF17789" "PF00797" ...
[1] "PMID"
chr [1:737868] "2591067" "3458201" "3610142" "8889549" "12477932" "14702039" "14760718" "15221005" "15461460" ...
[1] "PROSITE"
chr [1:1787] "PS50835" "PS00477" "PS00284" "PS01174" "PS50082" "PS00678" "PS50294" "PS51186" "PS50860" ...
[1] "REFSEQ"
chr [1:285470] "NM_130786" "NP_570602" "NM_000014" "NM_001347423" "NM_001347424" "NM_001347425" "NP_000005" ...
[1] "SYMBOL"
chr [1:66091] "A1BG" "A2M" "A2MP1" "NAT1" "NAT2" "NATP" "SERPINA3" "AADAC" "AAMP" "AANAT" "AARS1" "AAVS1" ...
[1] "UCSCKG"
chr [1:222852] "ENST00000596924.1" "ENST00000263100.8" "ENST00000600123.5" "ENST00000595014.1" ...
[1] "UNIPROT"
chr [1:32129] "P04217" "V9HWD8" "P01023" "P18440" "Q400J6" "F5H5R8" "A4Z6T7" "P11245" "A0A024R6P0" "P01011" ...
項目内の登録数が揃っていないのが分かるだろう。
ALIASもSYMBOLも遺伝子名の情報であるが、実際にkeys()で取り出して違いを見てみる。
alias <- keys(org.Hs.eg.db, "ALIAS")
symbol <- keys(org.Hs.eg.db, "SYMBOL")

登録数を見てみると明らかにALIASの方が多くの遺伝子名が登録されており、ALIASにはSYMBOLには登録されていない遺伝子名が存在することがわかる。
length(alias)
length(symbol)

alias, symbolの登録遺伝子数
SYMBOLの遺伝子名はALIASにも登録されているので、同じ遺伝子に異なる遺伝子名を付けているのではなく、ALIASの方が多くの遺伝子に遺伝子名を付けているということだ。
length(intersect(alias, symbol))
setdiff(symbol, alias)

aliasとsymbolの共通項
〇 遺伝子変換 select()/mapIDs()
上記までで確認した項目名などの情報を使って実際に遺伝子表記を変換する。
・select()
データベースのある項目から他の項目の情報を取得するにはselect()機能を使用する。selectだけだと他のパッケージのselectと機能名が重複していることがあるので、AnnotationDbi::select()と書くと間違い無い。
具体的には次のように記載する。
select(x = データベースオブジェクト,
keys = 変換したい遺伝子のベクトル,
keytype = keysに指定したベクトルの表記法,
colums = 変換先の項目)
試しにENSEMBL IDをSYMBOLとENTREZIDに変換してみる。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
select(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
columns = c("SYMBOL","ENTREZID"))

selectの出力
GENENAMEやGENETYPEの情報も遺伝子を理解するのに有用である。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
select(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
columns = c("SYMBOL","GENENAME","GENETYPE"))

selectの出力
select()機能の注意点として、一つのkeyに対して複数の情報が得られる場合である。
以下の例では1つのENSEMBL IDにmRNAのRefseq IDとproteinのRefseq IDがあり、指定したkeysの数よりも大きなdata.frameが返されてしまう。追加でデータ整形が必要になることが多い。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
select(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
columns = c("REFSEQ"))
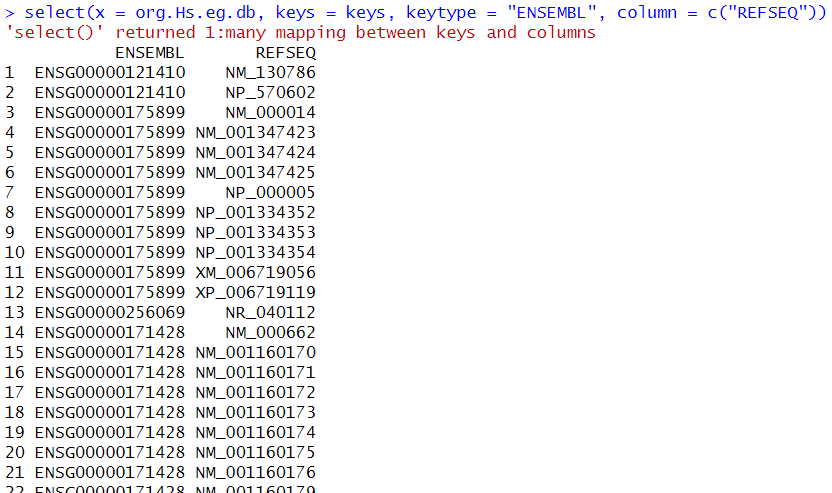
selectの出力
・mapIds()
似た機能にmapIds()があるが、こちらは項目を1つしか指定できない。
デフォルトの返り値はkeysが名前属性に入ったベクトルである。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
mapIds(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
column = "SYMBOL")

mapIdsの出力
しかし、mapIds()にはmultiVals=オプションがあり、1つのkeyに対して複数のhitがある場合の処理が選べる。
デフォルトではmultiVals="first"で、最初のhitが返されている。これであれば入力の要素数と出力の要素数が同じであるので扱いやすい場合もある。
multiVals="list"にすると1つのkeyに対する複数のhitをlistにまとめてくれる。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
mapIds(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
column = c("REFSEQ"),
multiVals = "list")

CharacterListというIRangesパッケージの専用クラスで返すこともできる。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
mapIds(x = org.Hs.eg.db,
keys = keys,
keytype = "ENSEMBL",
column = c("REFSEQ"),
multiVals = "CharacterList")

〇 Bimap objectを使った変換
org.生物種.eg.dbパッケージを読み込むと、「org.〇〇.egCHR」や「org.〇〇.egSYMBOL」のようなオブジェクトも読み込まれている。
これらは遺伝子IDと1つの項目の対応関係を保存したオブジェクトで、AnnotationDbiではBimap objectと名付けている。org.Hs.egSYMBOLなら、EntreZ IDと遺伝子名の対応情報である。
このオブジェクトを使うと、EntreZ IDから遺伝子名を簡単に変換できる。
オブジェクトを打ってみても中身は参照できないが、リスト化すると大体の中身がわかる。

オブジェクト名を打っても中身はわからない。
x <- as.list(org.Hs.egSYMBOL)

リスト化するとEntrez IDが名前属性に入った遺伝子名のリストが得られる
これをunlist()してやると名前付きベクトルが得られる。

名前属性にEnterZ IDがあることを利用すれば、簡単に情報が得られる。
xx[c(1,10,100)]

〇 clusterProfilerのbitr()機能
clusterProfilerパッケージのbitr()機能でもID変換が可能であるが、これも内部でAnnotationDbiを使用している。
引数名が違うぐらいでselect()機能と使い方は殆ど変わらない。
library(clusterProfiler)
genes <- head(keys(x = org.Hs.eg.db, keytype = "SYMBOL")) # デモ用遺伝子ベクトル
bitr(geneID = genes,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
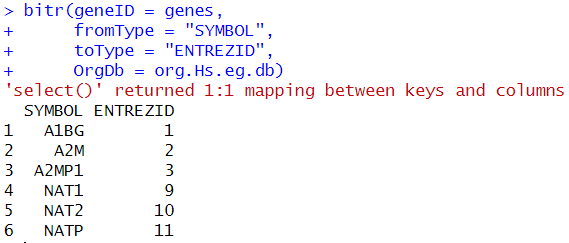
bitrの出力
ただデフォルト設定では、変換が旨くいかなかった遺伝子を結果から除いてしまう。
遺伝子を減らしたくない場合や、変換がうまくいかなかった遺伝子を知りたい場合は、drop=FALSEオプションをつけておくよい。
bitr(geneID = genes,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db,
drop=FALSE)
【 Ensdbパッケージ 】
Ensemblデータベースを基にした遺伝子データベース。
ヒトの物でも複数のversionのパッケージが提供されているが、genome versionやensembl versionの違いにより登録されている遺伝子数、転写産物数が違うようである。
# BiocManager::install("EnsDb.Hsapiens.v75")
library(EnsDb.Hsapiens.v75)
EnsDb.Hsapiens.v75

EnsDb.Hsapiens.v75 64102個の遺伝子、215647個の転写産物
# BiocManager::install("EnsDb.Hsapiens.v86")
library(EnsDb.Hsapiens.v86)
EnsDb.Hsapiens.v86

EnsDb.Hsapiens.v86 63970個の遺伝子、216741個の転写産物
項目確認
AnnotationDbiのkeytypes()機能で項目名が確認できる。
keytypes(EnsDb.Hsapiens.v86)
[1] "ENTREZID" "EXONID" "GENEBIOTYPE" "GENEID" "GENENAME"
[6] "PROTDOMID" "PROTEINDOMAINID" "PROTEINDOMAINSOURCE" "PROTEINID" "SEQNAME"
[11] "SEQSTRAND" "SYMBOL" "TXBIOTYPE" "TXID" "TXNAME"
[16] "UNIPROTID"
注意点は、ENSEMBLという項目名が無く、このデータベースではGENEIDがEnsembl IDである。
遺伝子変換
AnnotationDbiのselect()が上述と同様に使用可能である。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL")) # デモ用遺伝子ベクトル
AnnotationDbi::select(x = EnsDb.Hsapiens.v86,
keys = keys,
keytype = "GENEID",
columns = "ENTREZID")

【 bioMartパッケージ 】
おそらくこれが一番、多量の情報を取得できる。その分、欲しいデータまでたどり着くのが大変。ここではあくまで遺伝子名 ⇄ 遺伝子ID変換 に必要な情報のみ扱う。
bioMartはウェブ上からも同様のデータを取得することができる。ウェブサイト上部タブのBioMartから移行できる。
これからfiltersやattributesといった概念で情報の選別をしていくのだが、コマンドラインだと情報が膨大でわかりづらいのでブラウザーで感覚をつかんでおくと良いだろう。
togotvでもブラウザ操作が紹介されている。
# BiocManager::install("biomaRt")
library(biomaRt)
使用するMartの選択
listMarts()

db <- useMart(biomart = "ENSEMBL_MART_ENSEMBL") # useMart("ensembl")でも良いみたい。
db
Object of class 'Mart':
Using the ENSEMBL_MART_ENSEMBL BioMart database
No dataset selected.
このようにしてミラーサイトを指定することも可能。server errorが出る際にはこちらの方がいいかも。
db <- useEnsembl(biomart = "ENSEMBL_MART_ENSEMBL",
mirror = "asia",
dataset = "hsapiens_gene_ensembl")
データベースの選択
使用できるデータベース一覧を表示
head(listDatasets(db))
213種の生物種のデータベースが登録されている。

ヒトのデータベースを取得するには以下を実行する。
hg <- useDataset("hsapiens_gene_ensembl", mart = db)
項目確認
bioMartのブラウザでは、Filtersの画面で「欲しい遺伝子の制限 (特定の染色体番号や、queryとする遺伝子一覧など)」を行う。
一方、Rで情報取得する際のgetBM()機能では、values=引数でqueryの遺伝子を指定し、filters=引数でqueryとする遺伝子の表記法を指定する。attributes=引数で「欲しい項目 (遺伝子名や遺伝子IDなど)」を指定するところはブラウザと同様である。
getBM()機能のfilters=引数、attributes=引数で指定可能なtermを調べるには、listFilters()機能、listAttributes()機能が有用である。ただし選択可能なtermが膨大で、Rのコマンドの出力から探すよりブラウザで見る方が現実的である。
Attributesの方は、ホモログの情報も含んでおり、他の生物種の情報も入れると膨大な項目が得られる。
page = "feature_page"にするとまだ見やすいだろう。
listAttributes(mart = hg, page = "feature_page")
情報取得 getBM()
ENSEMBL IDをkeyにして、遺伝子名を取得する例を示す。デモ用に6つのEnsembl IDを用意した。
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL"))
keys
[1] "ENSG00000121410" "ENSG00000175899" "ENSG00000256069" "ENSG00000171428" "ENSG00000156006" "ENSG00000196136"
入力する遺伝子の表記はEnsembl ID。上記で確認した通りRでの指定名は"ensembl_gene_id"になる。
attributesには遺伝子名っぽい項目が幾つかあるので、試しにひとまず全て使用してみる。
実際の情報取得にはgetBM()機能を使用する。attributes=に欲しい項目、 mart=に使用するデータベース、filters=にvaluesに指定する遺伝子表記法, values=にqueryとなる遺伝子ベクトルを指定した。
getBM(attributes = c("ensembl_gene_id","hgnc_symbol","external_gene_name","wikigene_name"),
mart = hg,
filters = "ensembl_gene_id",
values = keys)

data.frameの出力が得られるので、適宜成型して使用できる。
ただし、ヒットしたものしか返ってこないのでvalues=に指定した全てがうまく変換されていなくても何のメッセージも出ない。また1つのEnsembl IDに遺伝子名が2つあったりと候補が複数あると行数が増える。

ちなみに、filters=やvalues=を指定せずにattributes=とmart=引数だけを使用すると、全遺伝子の指定attributesが取得可能である。

【 GTFファイルから対応表作り 】
RNAseq/scRNAseqのmappingを自分でやる時に、遺伝子のアノテーション情報ファイルのGTFファイルを扱う。GENCODEから取得したGTFファイルには既にEnsembl IDや遺伝子名の情報が入っている。
GTFファイルは単なるtableファイルなので頑張ればRで読み込めるが、9列目の情報は雑多で成型がめんどくさい。
rtracklayerを使えば列の区切りを認識してうまいこと読み込んでくれる。
BiocManager::install("rtracklayer")
library(rtracklayer)
GTFファイルの読み込み
gtf_filepath <- "gencode.v32.primary_assembly.annotation.gtf"
# GTFファイルは巨大なので読み込みには時間がかかる。
gtf <- rtracklayer::import(gtf_filepath) # readGFF()でもよい。
gtf_df <- as.data.frame(gtf)
gene_id列やgene_name列が既に作られている。
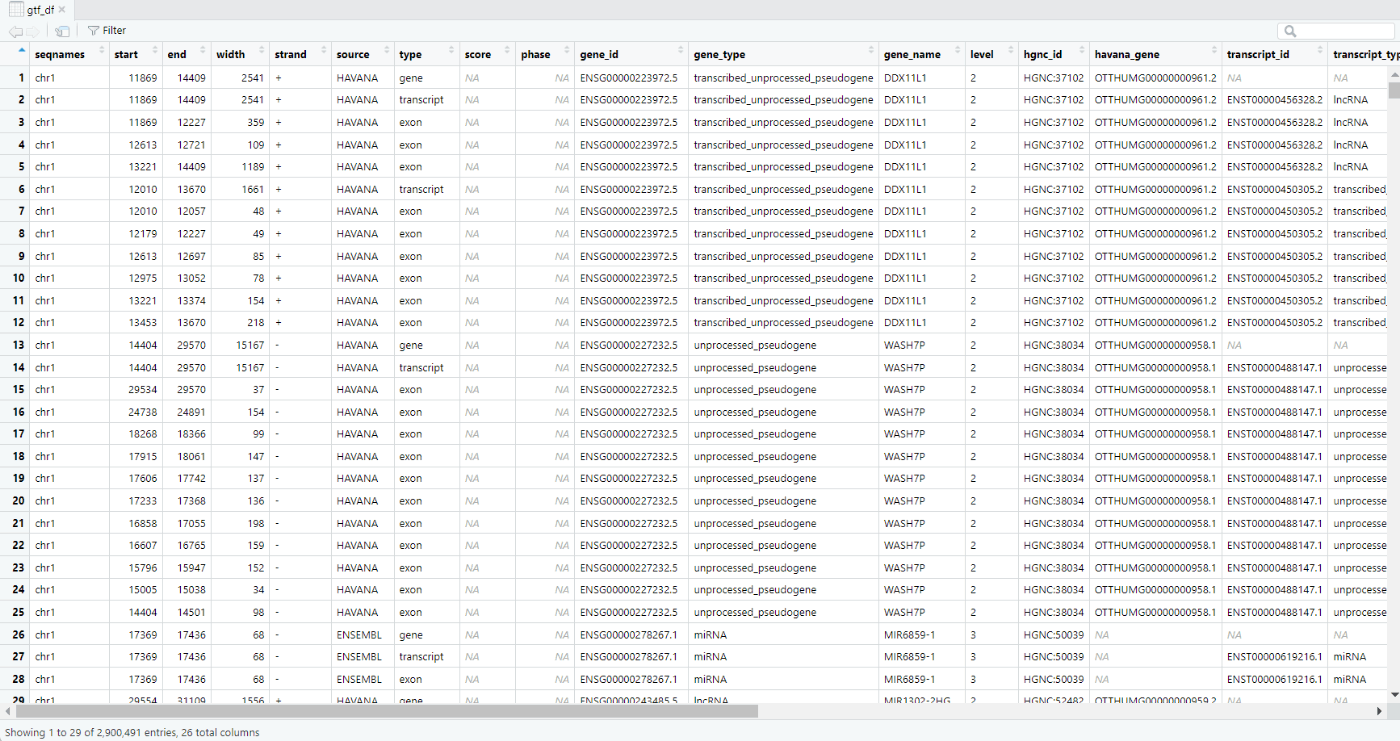
読み込んだGTFファイル
GTFファイルは転写産物レベルの情報も保持しているが、遺伝子レベルの情報に絞ればかなり情報が削減できる。
# 遺伝子IDと遺伝子名を抽出
gene_info <- gtf_df[, c("gene_id", "gene_name")]
# 重複を削除
gene_info_unique <- unique(gene_info)
こんな風に簡単に対応表が作成できた。

遺伝子IDのversion番号を除く
この例では「遺伝子ID.数字」という風にIDのversionが入っている。後の解析には不要なので削除しておく。
gene_info_unique$gene_id2 <-
stringr::str_split(gene_info_unique$gene_id, pattern = "\\.",simplify = TRUE)[,1]
ただし、versionを無くすと遺伝子IDに重複が生じる。
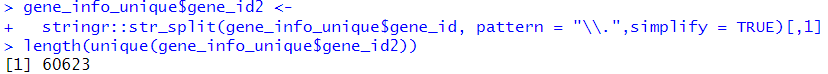
しかし、重複した遺伝子を見てみるとY遺伝子のものばかりで、不要だと思えば除いてもよいだろう。
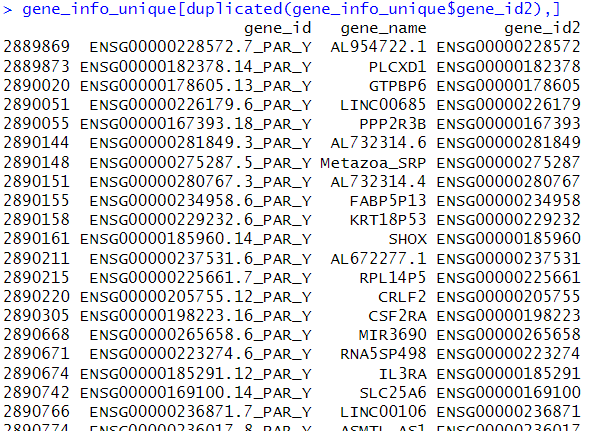
gene_info_unique <- gene_info_unique[!duplicated(gene_info_unique$gene_id2),]
rownames(gene_info_unique) <- gene_info_unique$gene_id2
【 MyGene 】
MyGeneは、遺伝子アノテーション情報を提供するオンラインリソースで、RやPythonから遺伝子の情報取得やIDの変換などを行うことができる。
BioGPSというWebサイト内でも使われているとのこと。
Web server上でアノテーション情報取得を行うことも可能。
Bioconductorからインストールできる。
BiocManager::install("mygene")
library(mygene)
アノテーションパッケージは、パッケージ内に遺伝子の情報が入ったものであったが、MyGeneパッケージは独自の遺伝子情報serverにアクセスして情報を取得する。
データベースのメタデータ
参考までにどんなデータを扱っているのか確認する場合は、以下のようにアクセスできる。
tmp <- MyGene()
meta <- metadata(tmp)
結果はメタデータのリストである。かなり豊富な情報が入っていることが分かる。生物種はヒト、マウスを含む9種類に対応している。

情報源も新しいものが使われているようだ。

遺伝子の情報取得 getGene() / getGenes()
keyとなる遺伝子を渡して、その遺伝子の情報を取得する機能。1つの遺伝子の場合はgetGene()機能、複数の遺伝子のベクトルではgetGenes()機能を使用する。
入力の遺伝子はEntrez IDかEnsembl IDを受け付ける。(遺伝子IDは生物種ごとに違うIDになっているので不要だが、species="human"のように生物種を指定可能。)
デモ用に6つのEnsembl IDを用意した。
library(org.Hs.eg.db)
keys <- head(keys(org.Hs.eg.db, keytype = "ENSEMBL"))

getGene()
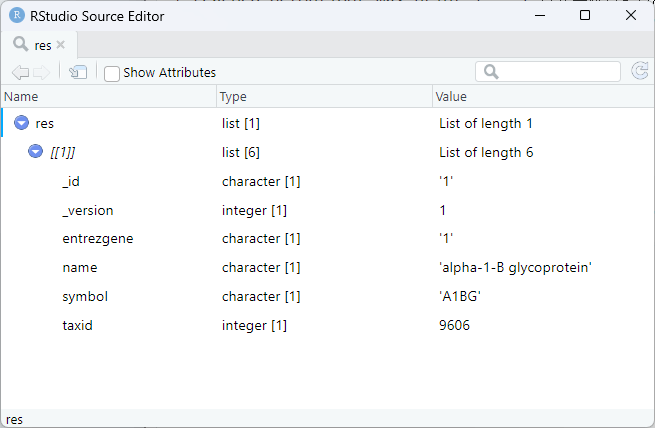
1つの遺伝子を引数に取る。返り値はリストである。
res <- getGene(keys[1])
getGenes()
複数の遺伝子を引数に取る。デフォルトの返り値はDataFrameというクラスで、通常のdata.frameと同様に扱える。

取得する項目の指定
どちらの機能もfields=引数で欲しい情報の項目を指定できる。デフォルトではfields = c("symbol", "name", "taxid", "entrezgene")である。fields=="all"とすると全項目が取得できる(146項目もあった。)
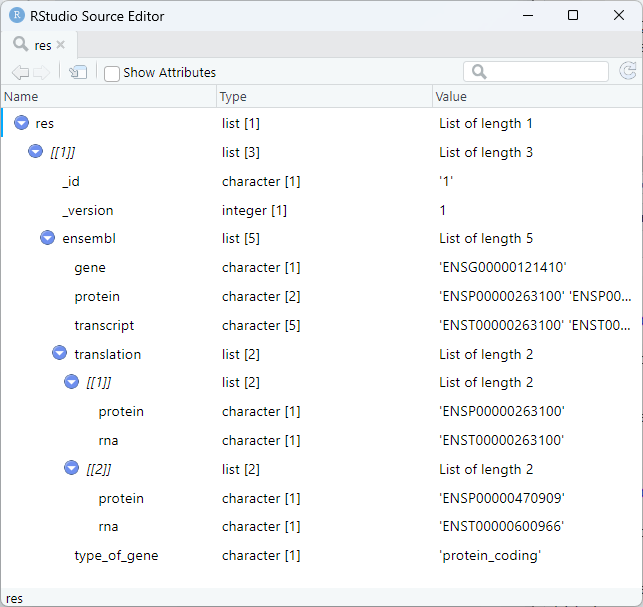
fields=引数に"ensembl"としていするとEnsemblデータベース内のgene、transcript、proteinなど複数の大項目で分かれて情報が取得される。
res <- getGene(keys[1], fields = c("ensembl"))
fields = c("ensembl")
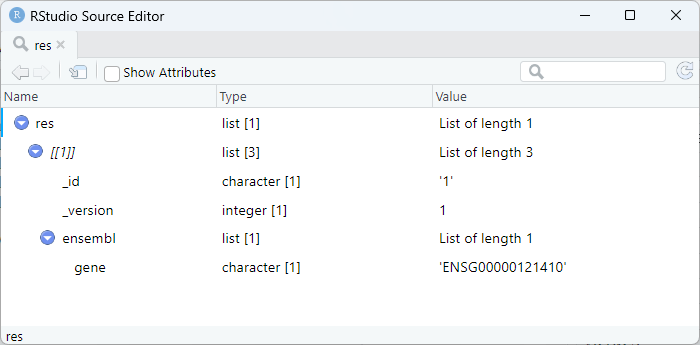
1つのデータベースの特定の分類の情報だけ欲しい場合は、.で区切って指定する。
res <- getGene(keys[1], fields = c("ensembl.gene"))
fields = c("ensembl.gene")
対応表作り
全てのEnsembl IDをSymbolやEntrez IDに簡単に変換できるように対応表を作る。
# 全てのEnsembl ID
keys <- keys(org.Hs.eg.db, keytype = "ENSEMBL")
geneTable <- getGenes(keys) # input遺伝子が多いときは1000個ずつ処理する
これを使えば簡単に遺伝子名などに変換可能である。

Ensembl IDの対応表
server versionの指定
getGene()/getGenes()機能にはmygene=引数がある。自分でMyGeneクラスを定義して渡すことが可能。

Rのhelpページを見るとbase.url=の指定がversion2のものになっているが、MyGeneのホームページを見るとversion3が出ている。
以下のようにserver versionを最新のものにして情報を取得することもできる。
mygene_v3 <- MyGene(base.url = "http://mygene.info/v3")
# version 3のserverで対応表作り
geneTable2 <- getGenes(keys, fields = c("symbol","entrezgene"), mygene = mygene_v3)
※ この例ではどちらのversionを使っても結果は同じ。
遺伝子情報の検索 `query()`/`queryMany()`
MyGeneサーバーに検索をかける機能。これであれば遺伝子名などからも検索可能。
本記事の用途には合わないので割愛する。
遺伝子名での検索
大文字小文字を区別していないようなので、ヒトの遺伝子表記ルールに従って全て大文字で遺伝子名を指定してもヒト以外の生物種の情報が返ってくる。species="human"のように生物種を指定した方が無難である。
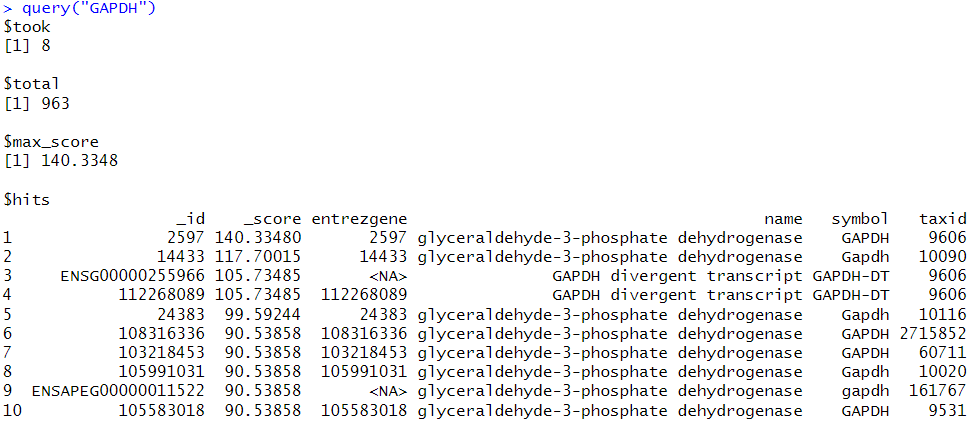
大文字で指定
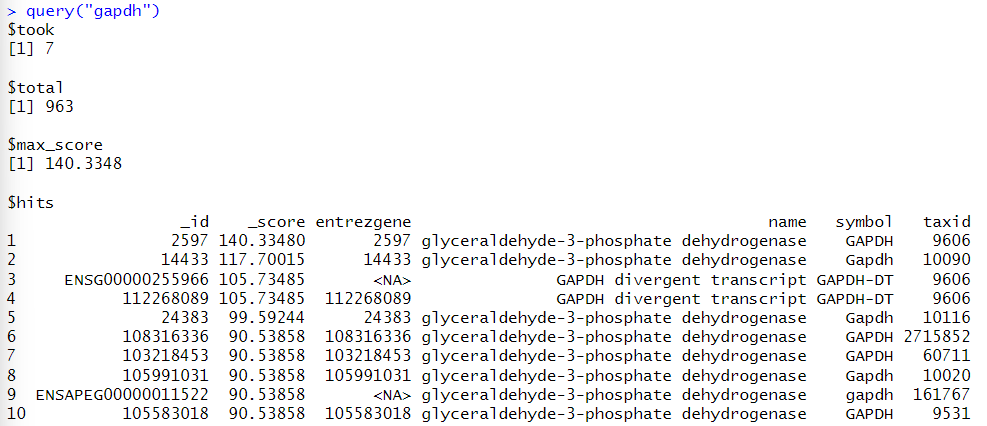
小文字で指定
【 異種生物間での変換 】
biomaRtパッケージ getLDS()
getLDS()機能を使って2つのdataset間でhomology mappingを行うことができる。
例として、ヒト遺伝子名からマウス遺伝子名に変換してみる。
library(biomaRt)
library(org.Hs.eg.db)
# デモ用にヒト遺伝子名を取得
keys <- head(keys(org.Hs.eg.db, keytype = "SYMBOL"))
ヒト、マウスそれぞれのMartオブジェクトを作成。
human <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
mouse <- useMart("ensembl", dataset = "mmusculus_gene_ensembl")
getLDS(attributes = c("hgnc_symbol"),
filters = "hgnc_symbol",
values = keys,
mart = human,
attributesL = c("mgi_symbol"),
martL = mouse,
uniqueRows=T)
(23年3月 記事作成時にはserver errorが出たため出力例は無し。)
Error: biomaRt has encountered an unexpected server error.
Consider trying one of the Ensembl mirrors (for more details look at ?useEnsembl)
23年9月 記事作成時から数か月経つが、mirror siteを変えても同じエラーが出る。

mirror="asia"

mirror="useast"
どうやらEnsembl versionを下げるとうまくいくらしく、host=引数を使ったmartの取得方法が紹介されていた。
https://support.bioconductor.org/p/9144001/
https://support.bioconductor.org/p/9143401/
確かにこの方法でヒト遺伝子名とマウス遺伝子名の対応表を作ることができた。
human <- useMart("ensembl", dataset = "hsapiens_gene_ensembl",
host = "https://dec2021.archive.ensembl.org/")
mouse <- useMart("ensembl", dataset = "mmusculus_gene_ensembl",
host = "https://dec2021.archive.ensembl.org/")
getLDS(attributes = c("hgnc_symbol"),
filters = "hgnc_symbol",
values = keys,
mart = human,
attributesL = c("mgi_symbol"),
martL = mouse,
uniqueRows=T)

Orthology.eg.db
Orthology mappingのためのパッケージ。ENTREZ IDを使うので、それ以外の遺伝子表記から開始する場合はいったんENTREZ IDに変換する必要がある。
例として、ヒト遺伝子名からマウス遺伝子名へ変換してみる。
# BiocManager::install("Orthology.eg.db")
library(Orthology.eg.db)
# 生物種ごとのパッケージも読み込んでおく。
library(org.Hs.eg.db) # BiocManager::install("org.Hs.eg.db")
library(org.Mm.eg.db) # BiocManager::install("org.Mm.eg.db")
# デモ用にヒト遺伝子名を取得
keys <- head(keys(org.Hs.eg.db, keytype = "SYMBOL"), 10)
# ENTREZ IDに変換
keys_ENTREZID <- mapIds(x = org.Hs.eg.db,
keys = keys,
keytype = "SYMBOL",
column = "ENTREZID")

遺伝子名をENTREZIDに変換
select()機能を使って変換を行う。keytype=に変換前の生物種、columns=に変換後の生物種名を指定する。
mapped <- select(Orthology.eg.db,
keys = keys_ENTREZID,
keytype = "Homo_sapiens",
columns = "Mus_musculus")
ヒトのENTREZ IDとマウスのENTREZ IDの対応表が得られる。

最後にENTREZ IDをマウス遺伝子名に変換する。(必要であれば)
keys_mSymbol <- mapIds(org.Mm.eg.db,
keys = as.character(na.omit(mapped$Mus_musculus)),
column = "SYMBOL",
keytype = "ENTREZID")

Discussion