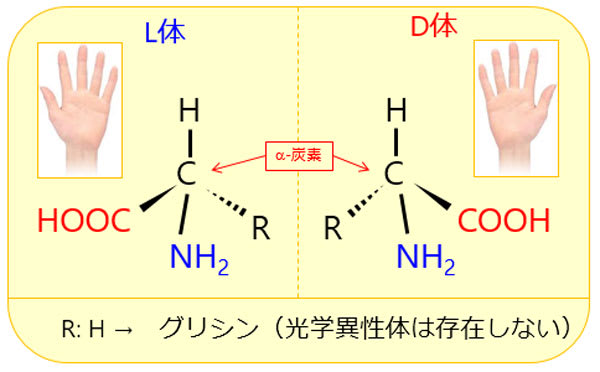
PyMOLで環状ペプチドの構造を出力する
PyMOLには標準でペプチドをbuildする機能が備わっています。
Macだとoptキーを押しながらアミノ酸配列を入力するだけです。
キーの割り当てがあればアミノ酸以外もbuild出来ます。
例えばopt+jにはアセチレンが割り当てられていますが、これはatom pick(原子の選択)が外れてしまうので、以降はペプチドの出力を続けることが出来ません。
下記はoptキーを押しながらHELPJPと入力することで出力したペプチドです。
途中がアセチレンなので最後のプロリンは出力されていません。(あと何故かロイシンも...)
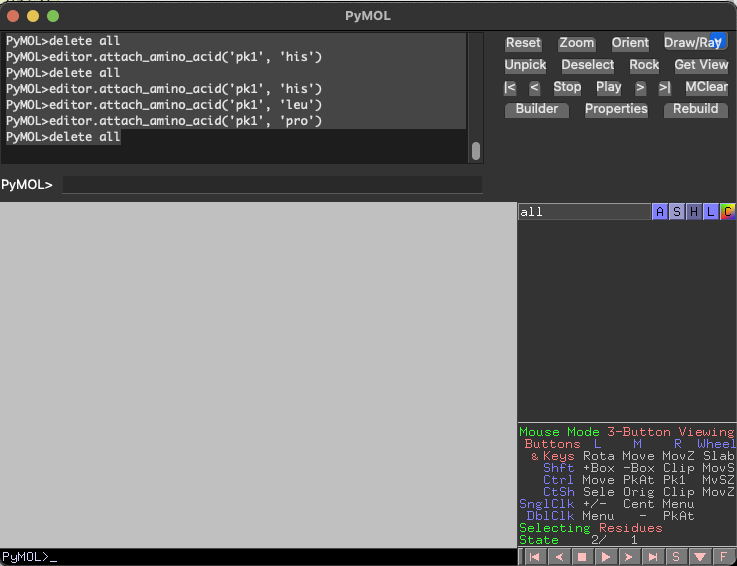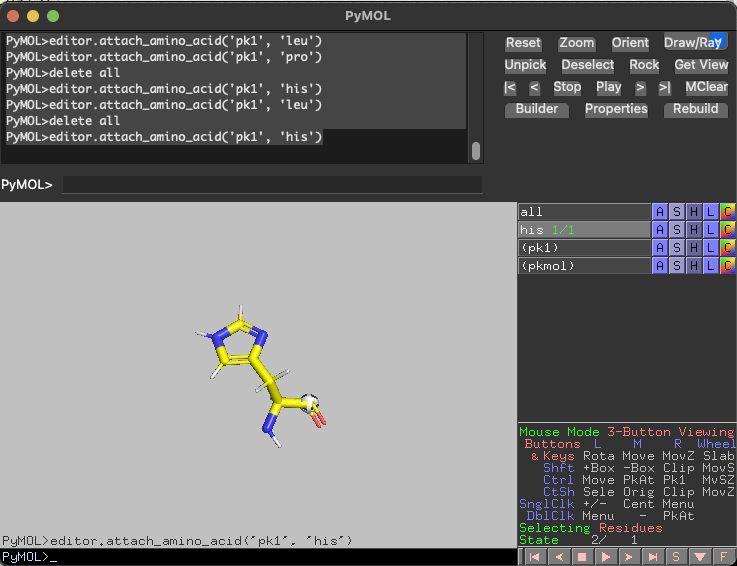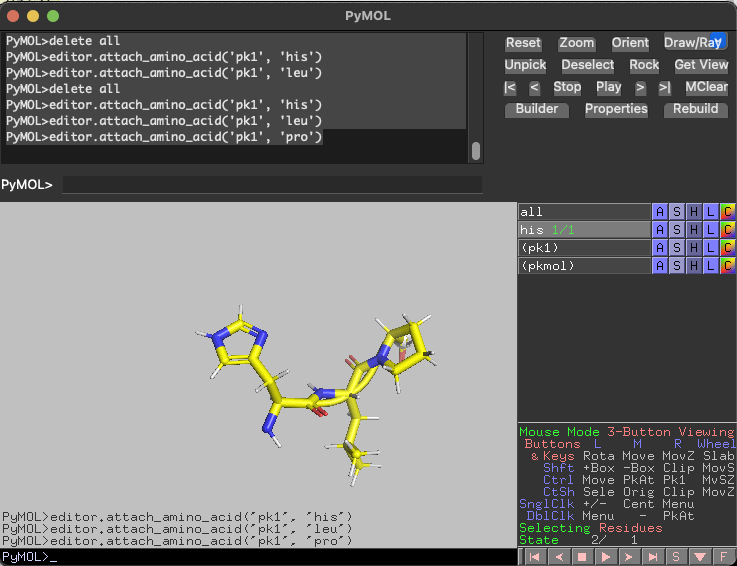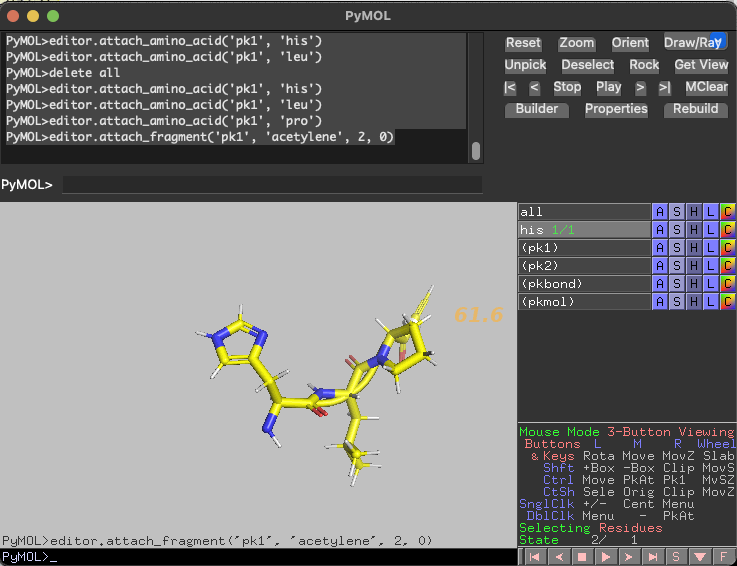
モチーフとしてはhelix, parallel beta sheet, antiparallel beta sheet(default)が用意されています。
最近ではPymolFoldというextentionsでESMFoldのAPIに配列を投げた予測構造を出力することもできます。(配列が外部サーバに投げられるので要注意)
こういった予測構造は予測スコアとともにfoldingまでしてくれるので大変優秀ですが、基本的に天然配列や直鎖に制限されています。
(加えて、原因はわかりませんが私の環境だとIとLのキーでアミノ酸が出力されません。ツールバーにはちゃんと割り当てが表示されてることは確認しています。)
というわけで、環状ペプチドを出力しようと言うのが本記事の内容です。
ついでにD型ペプチドにも対応しておきました。
スクリプトとインストール
下記のスクリプトを保存したディレクトリでPyMOLを起動して、コマンドをインストールします。
rdkitを使っているので、インストールをお忘れなく。
from rdkit import Chem
from rdkit.Chem import AllChem
from pymol import cmd
residues = {
'G': '[H]',
'A': '([H])(C)',
'V': '([H])(C(C)C)',
'L': '([H])(CC(C)C)',
'I': '([H])(C(C)CC)',
'M': '([H])(CCSC)',
'F': '([H])(CC1=CC=CC=C1)',
'W': '([H])(CC1=CNC2=C1C=CC=C2)',
'S': '([H])(CO)',
'C': '([H])(CS)',
'Y': '([H])(CC1=CC=C(C=C1)O)',
'N': '([H])(C(C(=O)N))',
'Q': '([H])(C(CC(=O)N))',
'D': '([H])(C(C(=O)O))',
'E': '([H])(C(CC(=O)O))',
'H': '([H])(C(C1=CNC=N1))',
'K': '([H])(CCCCN)',
'R': '([H])(CCCNC(=N)N)',
'T': '([H])([C@]([H])(O)C)',
'a': '(C)([H])',
'v': '((C(C)C)([H])',
'l': '(CC(C)C)([H])',
'i': '(C(C)CC)([H])',
'm': '(CCSC)([H])',
'f': '(CC1=CC=CC=C1)([H])',
'w': '(CC1=CNC2=C1C=CC=C2)([H])',
's': '(CO)([H])',
'c': '(CS)([H])',
'y': '(CC1=CC=C(C=C1)O)([H])',
'n': '(C(C(=O)N))([H])',
'q': '(C(CC(=O)N))([H])',
'd': '(C(C(=O)O))([H])',
'e': '(C(CC(=O)O))([H])',
'h': '(C(C1=CNC=N1))([H])',
'k': '(CCCCN)([H])',
'r': '(CCCNC(=N)N)([H])',
't': '([C@]([H])(O)C)([H])',
'g': '(C)(C)',
'J': '([H])(CS8)',
'j': '(CS8)([H])',
}
# aa format
aa = 'N[C@@]{X}C(=O)'
aaa = {k: aa.format(X=v) for k, v in residues.items()}
# add prolines and threonines
aaa['P'] = 'N1[C@@]([H])(CCC1)C(=O)'
aaa['p'] = 'N1[C@@](CCC1)([H])C(=O)'
# PepType
PepType = {
'0': 'Liner',
'1': 'Head to Tail',
'2': 'Symple Disulfide',
'3': 'Cystein Cyclization'
}
# PyMOL command
def peptide(seq, mode='1', name=None):
if name == None:
name = seq
# generate SMILES from fasta
sec = ''.join([aaa[aa] for aa in seq])
# Mode of Cyclyzation
if mode == '1':
smiles = 'N9' + sec[1:-4] + '9(=O)' # Head to Tail
elif mode == '2':
smiles = 'N[C@@]([H])(CS9)C(=O)' + sec + 'N[C@@]([H])(CS9)C(=O)O' # Symple Disulfide
elif mode == '3':
smiles = 'C9C(=O)' + sec + 'N[C@@]([H])(CS9)C(=O)N' # Cystein Cyclization
else :
smiles = sec + 'O' # Liner
# generate structure
mol = Chem.MolFromSmiles(smiles)
mol = Chem.AddHs(mol)
# Check if molecules are valid
if not mol:
raise ValueError('Invalid Peptide SMILES')
if AllChem.EmbedMolecule(mol, randomSeed=42) == -1:
# Try a different random seed
if AllChem.EmbedMolecule(mol, randomSeed=43) == -1:
raise ValueError("Embedding failed for peptide")
AllChem.UFFOptimizeMolecule(mol)
# Convert to PDB
pdb = Chem.MolToPDBBlock(mol)
# Load into PyMOL
cmd.read_pdbstr(pdb, name)
# cmd.remove(name)
cmd.show('sticks', name)
cmd.orient()
print(smiles)
cmd.extend('peptide', peptide)
run peptide.py
使い方
PyMOL consoleに配列を入力するとdefaultではLinerの構造を出力します。
オブジェクトに名前をつけたい時は, name=で定義してください。
consoleには一緒にSMILESを出力します。
peptide MERRYCHRISTMAS
peptide marrychristmas, name=d_santa
modeを使うことでHead to tail環化, ジスルフィド環化とチオエーテル環化に対応しました。
ジスルフィド環化は配列の先頭と末尾にシステインが付加されます。
チオエーテル環化はN末にアセチル、C末にシステインを付加して環化しています。ぺプチドリーム社が得意なやつです。
また、JとjにはそれぞれL型とD型のシステインでジスルフィド環化するように割り当てているので、配列の途中でジスルフィド環化させることもできます。Jとjがペアで存在していないとSMILESを生成できずにエラーが出るのでご注意を。
また、ジスルフィドがクロスするような配列には対応していません。
peptide headtail, mode=1
peptide DISULFIDE, mode=2
peptide peptidream, mode=3
peptide RGNJAYHKGQLVWJTYH, name=AJICAP
非天然アミノ酸には工夫が必要
天然配列とD型くらいならシンプルに構造を定義できますが、人工アミノ酸は多様性が低分子みたいなものなので配列で決定することは難しいです。
もしSMILESを自分で書ける知識やツールがあるなら、peptideコマンドで一緒に出力されるSMILESを修正してsmilesコマンドで再出力してみてください。
アミノ酸単位のSMILESは例えばL型では下記のようになっています。
print(aaa['Y'])
# N[C@@]([H])(CC1=CC=C(C=C1)O)C(=O)
L型の上記でL型のSMILESが出力されているので、H=([H]), R=(CC1=CC=C(C=C1)O)に相当しています。
 例えばこのSMILESを書き換えることで非天然アミノ酸を出力することができます。
例えばこのSMILESを書き換えることで非天然アミノ酸を出力することができます。
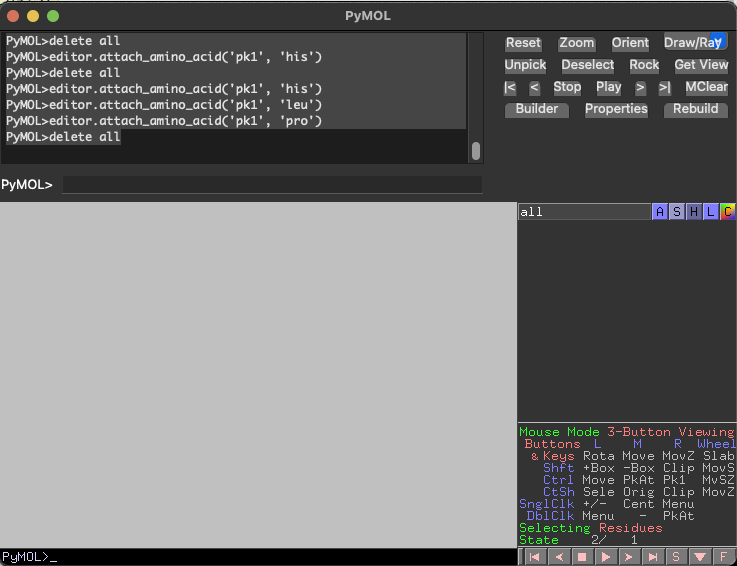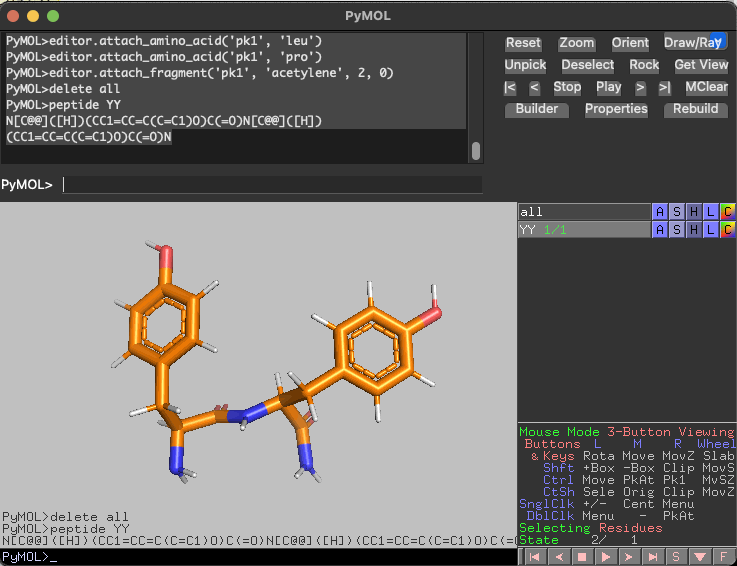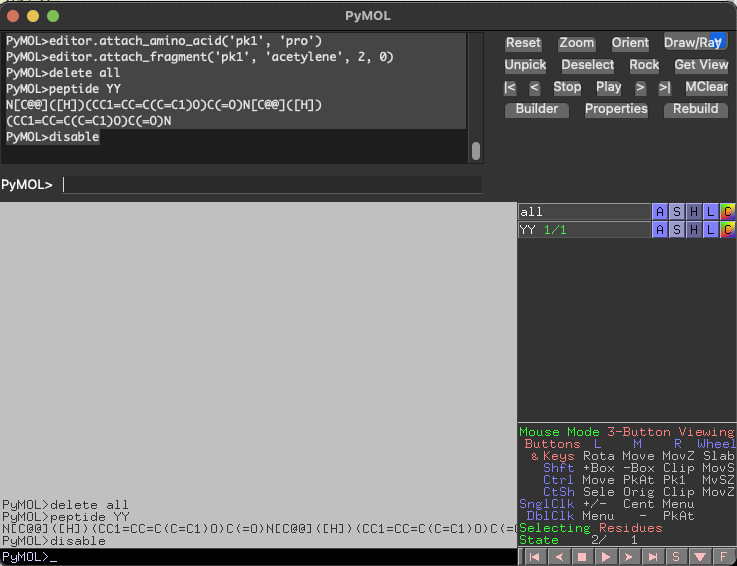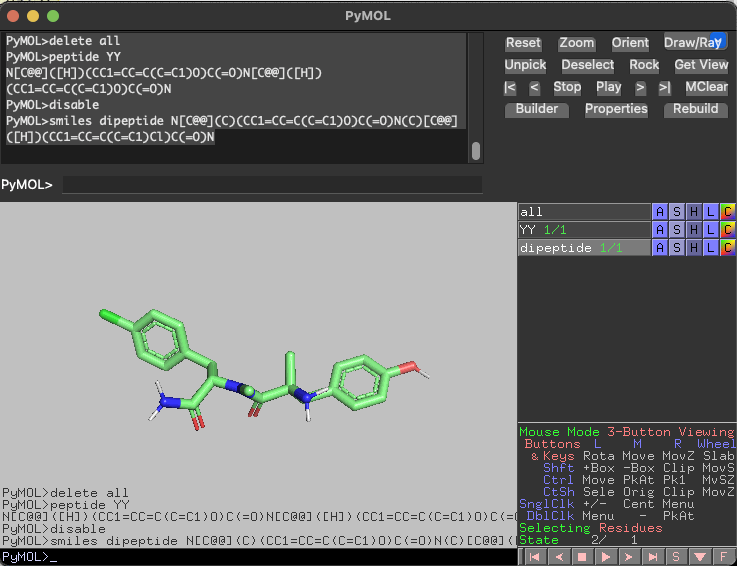
peptide YY
# NC[C@@](C)(CC1=CC=C(C=C1)O)C(=O)NC[C@@](C)(CC1=CC=C(C=C1)O)C(=O) # YY
smiles dipeptide N[C@@](C)(CC1=CC=C(C=C1)O)C(=O)N(C)[C@@]([H])(CC1=CC=C(C=C1)Cl)C(=O)N
# N[C@@](C)(CC1=CC=C(C=C1)O)C(=O)N(C)[C@@]([H])(CC1=CC=C(C=C1)Cl)C(=O)N # rewrited
もちろんPyMOLのbuilder機能を使って天然アミノ酸を非天然アミノ酸に描き換えてもいいと思います。
peptideコマンドで天然型のバックボーンを出力してからbuilderで修正、buildコマンドで再出力で綺麗な構造を再出力できます。
終わりに
非天然アミノ酸を色々いじって何が出来るようになるの?というのが次のステップです。
AlphaFold2では複合体の構造予測が出来るというのが初期にAFハックとしてTwitter(現X)に報告され、Natureにも引用されています。(私はこういうの大好きです)
しかし例に倣ってAlphaFold2で取り扱えるのは基本的に天然配列だけです。ではAF3は?
環状ペプチドは学習データが少なそうですが、どんな結果になるか楽しみだなと思います。
おわり。
Discussion