PyMOLでSMILESを扱う
この記事ではタイトルの通り、PyMOLでSMILESを扱う方法を紹介しています。
まどろっこしい説明が不要な方は、サンプルコードと使い方まですっ飛ばしてください。
(2024.Dec.16 追記)
OpenBabelに由来するエラーを見つけたのでrdkitのみで出力する方法に訂正しました。
また、描画した構造をbuildする際に元の構造が残っていると変な構造が出力されることがわかりました(多分PyMOLの使用?)。
そのため、ご自身で描いた構造は削除して正規化された構造のみ出力するように訂正しました。
SMILESとは
タンパクは基本的には20種類のアミノ酸で構成され、それぞれ固有のアミノ酸配列で表現されます。
DNAやRNAのような核酸はG・C・A・T(U)の4種類の塩基で構成され、それぞれ固有の核酸配列で表現されます。
これらの配列は便宜上1文字/残基or塩基で表されますが、実際には例えば1残基当たりでも水素を除いて5-15原子で構成されるため、タンパクや核酸(多くの場合RNA)の構造は非常に多様性に富んでいます。
一方の低・中分子はというと、自然界ですら使える原子から結合様式まで、高分子とは別の意味で多様性に富んでいます。
そのため、タンパクや核酸のような少ないパターンで表現することが難しいため、原子や結合自体を個別に表現することが多いです。
SMILESはこれら低・中分子の分子構造を記述するポピュラーな方法の一つです。
例えばプロパンはCCC、ピペラジンはC1NCCNC1のように記述します。
ここでは記述方法については取り扱いませんが、勘の言い方は複雑な分子だと複数の記述が出来そうと気付くかもしれません。
実際に複数の書き方が出来てしまうので、取り扱うソフトウェアの中でルールが決まっていることが多いです。
PyMOLでSMILESを扱うには
PyMOLはFASTAには対応していますが、SMILESには対応していません。
そのため、PyMOLと同じくPythonで動作するrdkitやopenbabelを使って、単なる文字列のSMILESをPyMOLで扱えるMOLファイルに相互変換します。
例えば前述のピペラジンは、
# import library
from rdkit import Chem, Draw
# definition of piperazine
smiles = 'C1NCCNC1'
# SMILES to mol file
mol = Chem.MolFromSmiles(smiles)
# Drawing mol
Draw.MolToImage(mol)
Drawのメソッドはjupyterなどで分子構造を確認するためのもので、PyMOLに出力するときは要りません。
反対にmolファイルをSMILESに変換するには、
Chem.MolToSmiles(mol)
この辺が基本です。
サンプルコード
細かい説明は省きますが(え?)、下記のスクリプトを保存してPyMOLで実行することで、SMILESをPyMOLで扱うためのいくつかのコマンドを import出来ます。
PyMOLと同じ仮想環境にrdkitとopenbabelをインストールしておくことをお忘れなく。
from rdkit import Chem
from rdkit.Chem import AllChem
from pymol import cmd
def smiles(arg_string):
# 引数をスペースで分割
args = arg_string.split()
# 最低限の引数チェック
if len(args) < 2:
print("エラー: コマンドには少なくとも名前とSMILES文字列が必要です。")
return
name = args[0]
smile = args[1]
# 必要に応じてpH引数を取得
pH = None
if len(args) > 2:
try:
pH = float(args[2])
except ValueError:
print(f"エラー: pH値 '{args[2]}' は有効な数値ではありません。")
return
if pH is not None:
smile = get_smi_with_pH(smile, pH)
mol = Chem.MolFromSmiles(smile)
if mol is None:
print(f"エラー: SMILES文字列 '{smile}' から分子を生成できませんでした。")
return
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
pdb_block = Chem.MolToPDBBlock(mol)
if pdb_block is None:
print("エラー: 分子のPDBブロックを生成できませんでした。")
return
cmd.read_pdbstr(pdb_block, name)
cmd.hide(f'({name} and hydro and (elem C extend 1))')
# コマンドをPyMOLに拡張
cmd.extend('smiles', smiles)
def printsmiles(selection="sele"):
# 一時SDFファイル名
tmp = "./tmp_molecule.sdf"
# PyMOLから選択された分子をSDF形式で保存
cmd.save(tmp, selection)
# rdkitで分子をロード、正規化
suppl = Chem.SDMolSupplier(tmp)
mol = next(suppl) if suppl and len(suppl) > 0 else None
if mol is None:
print("Error: Failed to load the molecule from the SDF file.")
return
smiles = Chem.MolToSmiles(mol)
# SMILES形式に変換し、オブジェクト名を追加して出力する
object_name = cmd.get_object_list(selection)[0]
print(f"SMILES:\n{object_name}\t{smiles}")
# 一時ファイルを削除
os.remove(tmp)
# コマンドをPyMOLに拡張
cmd.extend("printsmiles", printsmiles)
def build(selection="sele"):
# 一時SDFファイル名
tmp = "./tmp_molecule.sdf"
# PyMOLから選択された分子をSDF形式で保存
cmd.save(tmp, selection)
# rdkitで分子をロード、正規化
suppl = Chem.SDMolSupplier(tmp)
mol = next(suppl) if suppl and len(suppl) > 0 else None
if mol is None:
print("Error: Failed to load the molecule from the SDF file.")
return
smiles = Chem.MolToSmiles(mol)
# SMILES形式に変換し、オブジェクト名を追加して出力する
object_name = cmd.get_object_list(selection)[0]
print(f"SMILES:\n{object_name}\t{smiles}")
# SMILESから立体構造を生成
mol = Chem.MolFromSmiles(smiles)
if mol is None:
print(f"エラー: SMILES文字列 '{smiles}' から分子を生成できませんでした。")
return
mol = Chem.AddHs(mol)
AllChem.EmbedMolecule(mol)
AllChem.UFFOptimizeMolecule(mol)
pdb_block = Chem.MolToPDBBlock(mol)
if pdb_block is None:
print("エラー: 分子のPDBブロックを生成できませんでした。")
return
# PyMOLに立体構造を読み込み
cmd.remove(object_name)
cmd.read_pdbstr(pdb_block, object_name)
cmd.hide(f'({object_name} and hydro and (elem C extend 1))')
# 一時ファイルを削除
os.remove(tmp)
print(f"{object_name}の立体構造がPyMOLにロードされました。")
# コマンドをPyMOLに拡張
cmd.extend('build', build)
上記のスクリプトを保存したディレクトリでPyMOLを起動して、PyMOLのコマンドラインからスクリプトを実行します。
run smiles.py
これでコマンドのインストールは完了です。
使い方
SMILESを分子構造に変換するコマンドは下記の通りです。
試しに複雑な分子の代表格であるハリコンドリンを出力してみましょう。
ハリコンドリンのSMILESはPubchemから持ってきました。
2D structureが正常に出力されてませんね。。。構造の複雑さを物語っています。これをPyMOLに出力するには、PyMOLのコマンドラインで下記を実行します。
smiles halichondrin C[C@@H]1C[C@@H]2CC[C@H]3C(=C)C[C@@H](O3)CC[C@]45C[C@@H]6[C@H](O4)[C@H]7[C@@H](O6)[C@@H](O5)[C@@H]8[C@@H](O7)CC[C@@H](O8)CC(=O)O[C@@H]9[C@H]([C@H]3[C@H](C[C@@H]4[C@H](O3)C[C@@]3(O4)C[C@H]4[C@@H](O3)[C@H](C[C@]3(O4)C[C@@H]([C@H]4[C@@H](O3)C[C@H](O4)[C@H](C[C@H](CO)O)O)C)C)O[C@H]9C[C@H](C1=C)O2)C

注意点が1点あります。
通常のPyMOLのコマンドは2つ以上の引数をとる場合に,(カンマ)で区切りますが、このコマンドはスペースで区切っています。理由は...考えてみてください。
次に、出力したハリコンドリンのSMILESを下記のコマンドでPyMOLのコンソールに出力してみましょう。
printsmiles halichondrin
# SMILES:
# halichondrin C[C@@H]1C[C@@H]2CC[C@@H]3O[C@H](CC3=C)CC[C@]34O[C@H]5[C@H](O[C@@H]6[C@H]5O[C@@H]5[C@H](O[C@H](CC5)CC(=O)O[C@@H]5[C@H]([C@@H]7O[C@H]8[C@H](O[C@]9(O[C@@H]%10[C@@H](O[C@@]%11(O[C@@H]%12[C@@H](O[C@@H](C%12)[C@@H](O)C[C@@H](O)CO)[C@H](C%11)C)C[C@@H]%10C)C9)C8)C[C@@H]7O[C@H]5C[C@@H](O2)C1=C)C)[C@@H]6O3)C4
printsmilesの引数はオブジェクト名を指定しますが、デフォルトでは(sele)を出力します。
これの何が便利かというと、立体構造をキレイに再出力できます。
こちらで紹介したようにPyMOLは分子描画ソフトとしても使えますが、ChemDrawやMarvinよりも細かい調整が難しいので複雑な分子はキレイにしてから確認する必要があります。
描いてみた構造のSMILESをprintsmilesコマンドで出力してから、smilesコマンドで構造を再出力すると、ぐちゃぐちゃだった構造がキレイな形で再出力できます。
このprintsmiles→smilesの二度手間をひとまとめにしたのがbuildコマンドです。
build [object_name]
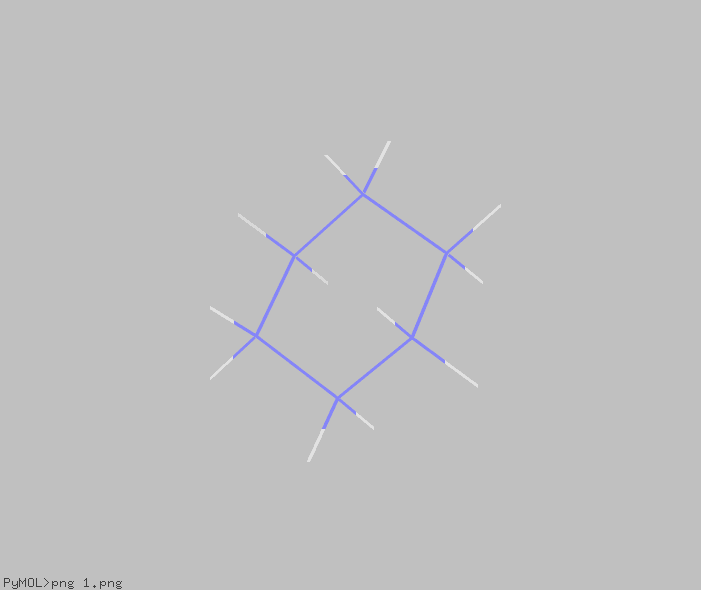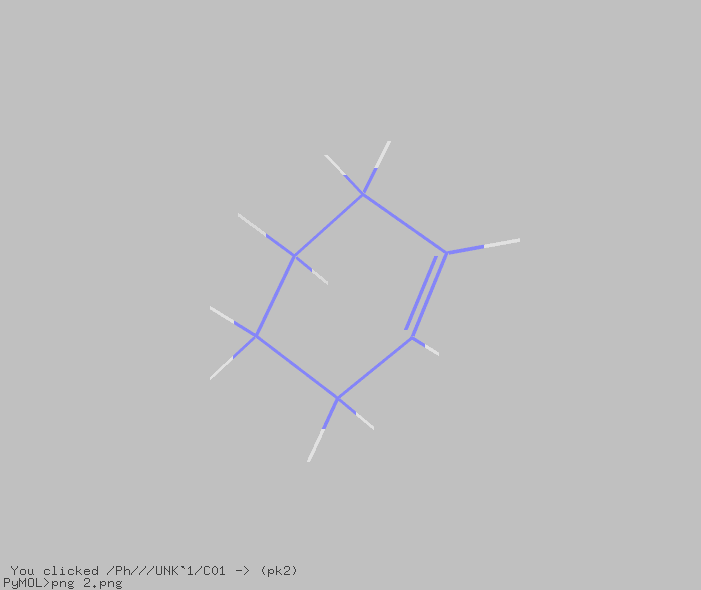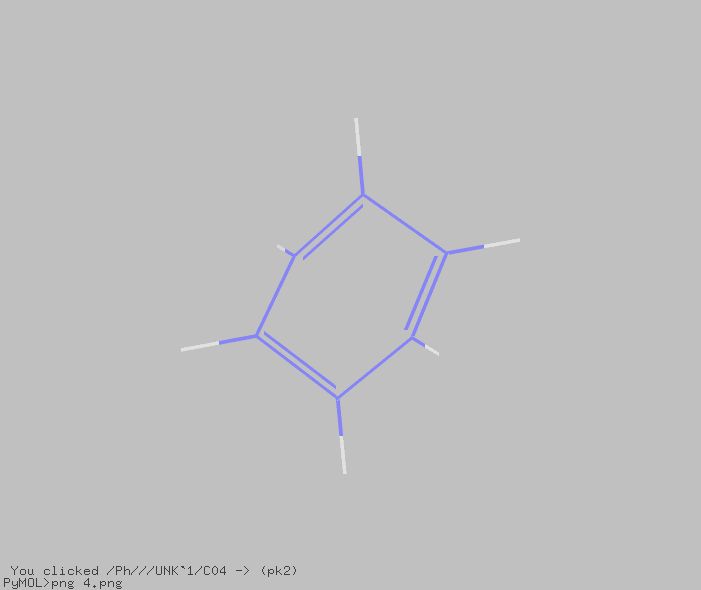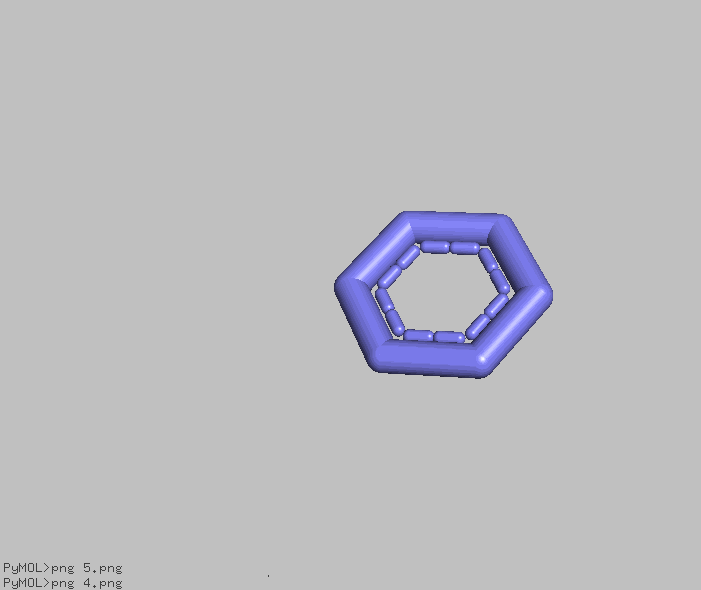
builderで描画した変な形の構造式を正規化することができました。
rdkitやopenbabelを使うことで、PyMOLの中だけで色んな変換が出来るようになります。
Discussion