GeMoMa: リファレンスの遺伝子構造情報などから相同性に基づき対象種の遺伝子を予測してアノテーションする

GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data
イントロンの位置の保存性とRNAseqのデータを利用した相同性に基づく遺伝子予測
はじめに
GeMoMa[1]はホモログ(相同遺伝子)活用したゲノムアノテーションツールで、リファレンスゲノム配列と既知のアノテーション情報から新規ゲノム配列の遺伝子構造を予測することができます。
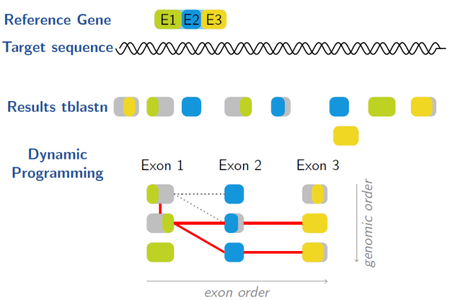
Schema of GeMoMa algorithm
Augustus[2]といったab-initioベースのアプローチは広範なトレーニングステップが必要であったり、RNAseqのデータを利用する場合でも限定的な利用に限る場合が多いです。また、低発現領域のExon-Intronの識別は非常に困難です。
一方でホモログベースのアプローチは、既存のモデル生物のアノテーション情報を利用するため、多少遠縁な系統にも適用が可能です。また、RNAseqのデータが無い生物種でもホモログ情報を主軸に予測を行うため限られたデータで遺伝子機能アノテーションを実行可能です。
GeMoMaの流れ
GeMoMaのパイプライン処理の基本的構造は以下の通りです。詳細は各工程の章に記載しています。
- Extract RNA-seq evidence(ERE)
- CheckIntrons
- DenoiseIntrons
- Extractor
- External Search (mmseqs2, tblastn)
- Gene Model Mapper (GeMoMa)
- GeMoMa Annotation Filter (GAF)
- Annotation Finalizer
記事の後半に記載していますがGeMoMaPipelineを使えば一括実行が可能です。
実行環境の構築
実行要件としてJava v1.8以降が必要となります。また、相同性検索が含まれている関係でblastかmmseqsが必要です。まずは仮想環境を構築してツールをインストールしていきます。
# フォルダの作成
mkdir ref output bam
mkdir GeMoMa; cd GeMoMa
mkdir env
# 仮想環境の作成
mamba create -p $PWD/env/gemoma -c conda-forge -c bioconda \
python=3.12 \
gemoma=1.9 \
star=2.7.11b \
fastp=0.23.4 \
pigz=2.8 \
aria2=1.36 \
sra-tools=3.1.1 \
samtools=1.21 \
seqkit=2.8.2
mamba create -p $PWD/env/trinity -c conda-forge -c bioconda \
kmer-jellyfish=2.3.1 \
salmon=1.10.3 \
bowtie2=2.5.4 \
samtools=1.21 \
trinity=2.15.1 \
STAR=2.7.11 \
fastq-pair
GeMoMa USAGE
Parameters of tool "GeneModelMapper" (GeMoMa, version: 1.9):
s - search results (The search results, e.g., from tblastn or mmseqs, type = tabular) = null
t - target genome (The target genome file (FASTA), i.e., the target sequences in the blast run. Should be in IUPAC code, type = fasta,fas,fa,fna,fasta.gz,fas.gz,fa.gz,fna.gz) = null
c - cds parts (The query CDS parts file (protein FASTA), i.e., the CDS parts that have been searched in the target genome using for instance BLAST or mmseqs, type = fasta,fa,fas,fna) = null
a - assignment (The assignment file, which combines CDS parts to proteins, type = tabular, OPTIONAL) = null
The following parameter(s) can be used zero or multiple times:
i - introns (Introns (GFF), which might be obtained from RNA-seq, type = gff,gff3) = null
r - reads (if introns are given by a GFF, only use those which have at least this number of supporting split reads, valid range = [1, 2147483647], default = 1) = 1
splice - splice (if no intron is given by RNA-seq, compute candidate splice sites or not, default = true) = true
The following parameter(s) can be used zero or multiple times:
coverage - coverage (experimental coverage (RNA-seq), range={UNSTRANDED, STRANDED}, default = UNSTRANDED) = UNSTRANDED
Parameters for selection "UNSTRANDED":
coverage_unstranded - coverage_unstranded (The coverage file contains the unstranded coverage of the genome per interval. Intervals with coverage 0 (zero) can be left out., type = bedgraph) = null
Parameters for selection "STRANDED":
coverage_forward - coverage_forward (The coverage file contains the forward coverage of the genome per interval. Intervals with coverage 0 (zero) can be left out., type = bedgraph) = null
coverage_reverse - coverage_reverse (The coverage file contains the reverse coverage of the genome per interval. Intervals with coverage 0 (zero) can be left out., type = bedgraph) = null
g - genetic code (optional user-specified genetic code, type = tabular, OPTIONAL) = null
sm - substitution matrix (optional user-specified substitution matrix, type = tabular, OPTIONAL) = null
go - gap opening (The gap opening cost in the alignment, default = 11) = 11
ge - gap extension (The gap extension cost in the alignment, default = 1) = 1
m - maximum intron length (The maximum length of an intron, default = 15000) = 15000
sil - static intron length (A flag which allows to switch between static intron length, which can be specified by the user and is identical for all genes, and dynamic intron length, which is based on the gene-specific maximum intron length in the reference organism plus the user given maximum intron length, default = true) = true
intron-loss-gain-penalty - intron-loss-gain-penalty (The penalty used for intron loss and gain, default = 25) = 25
rf - reduction factor (Factor for reducing the allowed intron length when searching for missing marginal exons, valid range = [1, 100], default = 10) = 10
e - e-value (The e-value for filtering blast results, default = 100.0) = 100.0
ct - contig threshold (The threshold for evaluating contigs, valid range = [0.0, 1.0], default = 0.4) = 0.4
h - hit threshold (The threshold for adding additional hits, valid range = [0.0, 1.0], default = 0.9) = 0.9
o - output (critierium to determine the number of predictions per reference transcript, range={STATIC, DYNAMIC}, default = STATIC) = STATIC
Parameters for selection "STATIC":
p - predictions (The (maximal) number of predictions per transcript, default = 10) = 10
Parameters for selection "DYNAMIC":
f - factor (a prediction is used if: score >= factor*Math.max(0,bestScore), valid range = [0.0, 1.0], default = 0.8) = 0.8
selected - selected (The path to list file, which allows to make only a predictions for the contained transcript ids. The first column should contain transcript IDs as given in the annotation. Remaining columns can be used to determine a target region that should be overlapped by the prediction, if columns 2 to 5 contain chromosome, strand, start and end of region, type = tabular,txt, OPTIONAL) = null
as - avoid stop (A flag which allows to avoid (additional) pre-mature stop codons in a transcript, default = true) = true
approx - approx (whether an approximation is used to compute the score for intron gain, default = true) = true
pa - protein alignment (whether a protein alignment between the prediction and the reference transcript should be computed. If so two additional attributes (iAA, pAA) will be added to predictions in the gff output. These might be used in GAF. However, since some transcripts are very long this can increase the needed runtime and memory (RAM)., default = true) = true
prefix - prefix (A prefix to be used for naming the predictions, default = ) =
tag - tag (A user-specified tag for transcript predictions in the third column of the returned gff. It might be beneficial to set this to a specific value for some genome browsers., default = mRNA)= mRNA
v - verbose (A flag which allows to output a wealth of additional information per transcript, default = false) = false
timeout - timeout (The (maximal) number of seconds to be used for the predictions of one transcript, if exceeded GeMoMa does not output a prediction for this transcript., valid range = [0, 604800], default = 3600) = 3600
sort - sort (A flag which allows to sort the search results, default = false) = false
ru - replace unknown (Replace unknown amino acid symbols by X, default = false) = false
Score - Score (A flag which allows to do nothing, re-score or re-align the search results, range={Trust, ReScore, ReAlign}, default = Trust) = Trust
outdir - The output directory, defaults to the current working directory (.) = .
At least one parameter has not been set (correctly).
At least parameter "search results" has not been set to a valid value.
リファレンスとする近縁種のタンパク配列(PROTAINS.fna)と遺伝子情報(.gff)の準備をします。
先行研究では以下の6種を対象にアユのドラフトゲノムとのシンテニー解析を実施していました。
- メダカ: Oryzias latipes
- ノーザンパイク: Esox lucius
- レインボートラウト: Oncorhynchus mykiss
- タイセイヨウサケ: Salmo salar
- ゼブラフィッシュ: Danio rerio
- スポッテッドガー: Lepisosteus oculatus
今回の遺伝子構造アノテーションにも同様の種を使用してみます。
各種のゲノム情報とアノテーション情報のEnsemblのリンクは以下を参照ください。
| Species Name | Name | Ensembl Genome Link |
|---|---|---|
| Esox lucius | fEsoLuc1.pri | https://asia.ensembl.org/Esox_lucius/Info/Index |
| Oncorhynchus mykiss | USDA_OmykA_1.1 | https://asia.ensembl.org/Oncorhynchus_mykiss/Info/Index |
| Salmo salar | Ssal_v3.1 | https://asia.ensembl.org/Salmo_salar/Info/Index |
| Oryzias latipes | ASM223467v1 | https://asia.ensembl.org/Oryzias_latipes/Info/Index |
| Danio rerio | GRCz11 | https://asia.ensembl.org/Danio_rerio/Info/Index |
塩基配列ファイルをダウンロードします。
cd ref
# array
genomes=(
https://ftp.ensembl.org/pub/release-113/fasta/esox_lucius/dna/Esox_lucius.fEsoLuc1.pri.dna_sm.toplevel.fa.gz
https://ftp.ensembl.org/pub/release-113/fasta/oncorhynchus_mykiss/dna/Oncorhynchus_mykiss.USDA_OmykA_1.1.dna_sm.toplevel.fa.gz
https://ftp.ensembl.org/pub/release-113/fasta/salmo_salar/dna/Salmo_salar.Ssal_v3.1.dna_sm.toplevel.fa.gz
https://ftp.ensembl.org/pub/release-113/fasta/oryzias_latipes/dna/Oryzias_latipes.ASM223467v1.dna_sm.toplevel.fa.gz
https://ftp.ensembl.org/pub/release-113/fasta/danio_rerio/dna/Danio_rerio.GRCz11.dna_sm.primary_assembly.fa.gz
)
# DL
for genome in ${genomes[@]};do
aria2c -x 16 -ctrue ${genome}
done
遺伝子定義ファイルをダウンロードします。
cd ref
# array
gffs=(
https://ftp.ensembl.org/pub/release-113/gff3/esox_lucius/Esox_lucius.fEsoLuc1.pri.113.chr.gff3.gz
https://ftp.ensembl.org/pub/release-113/gff3/oncorhynchus_mykiss/Oncorhynchus_mykiss.USDA_OmykA_1.1.113.chr.gff3.gz
https://ftp.ensembl.org/pub/release-113/gff3/salmo_salar/Salmo_salar.Ssal_v3.1.113.chr.gff3.gz
https://ftp.ensembl.org/pub/release-113/gff3/oryzias_latipes/Oryzias_latipes.ASM223467v1.113.chr.gff3.gz
https://ftp.ensembl.org/pub/release-113/gff3/danio_rerio/Danio_rerio.GRCz11.113.chr.gff3.gz
)
# DL
for gff in ${gffs[@]};do
aria2c -x 16 -ctrue ${gff}
done
GeMoMaの下準備
NCBIにあるアユのRNAseqデータでライブラリ調整キットの情報がはっきりしているのはSRR1533707 ~ SRR1533716のみでした。発現パターンが異なる複数の配列データがあると望ましいとは思います。ですが、データの充実度合いに依存するので無理に追加データを取る必要は無いと思います。
こちらをデータクリーニングします。最終的なデータは対象種のゲノムにRNAseqのデータをマップしたBamファイルです。
-
RNAseqのデータをTrinityでDe novo assemblyしてcontig配列を取得
De novo assemblyにより構築されるContigはRNAseqの中である程度のカバレッジを持つ領域の配列が利用されるので、マイナーなエラー配列や転写ミスなどの割合が低いノイジーな配列の影響が少ないリファレンス配列を作成できます。
-
STARを使ってRNAseqのデータをContig配列にマッピング
STARを使用してContig配列にRNAseqのReadをマッピングします。
-
Contig配列にマップされた配列を回収
SamtoolsでContig配列にマップされた配列を回収することでノイジーな配列を除去したRNAseqの配列データを得ることができます。
-
ペアとなる配列データのみとなるようにフィルタリング
Paired-Endのデータとして扱うので、ペア構造を維持している配列のみのFastqファイルとなるようfastq_pairでフィルタリングします。
-
対象種のゲノム配列に再度マップしてBamファイルを作成
再度、STARを使用して対象種のゲノム配列にフィルタリング後のゲノム配列をマッピングしたら終了です。
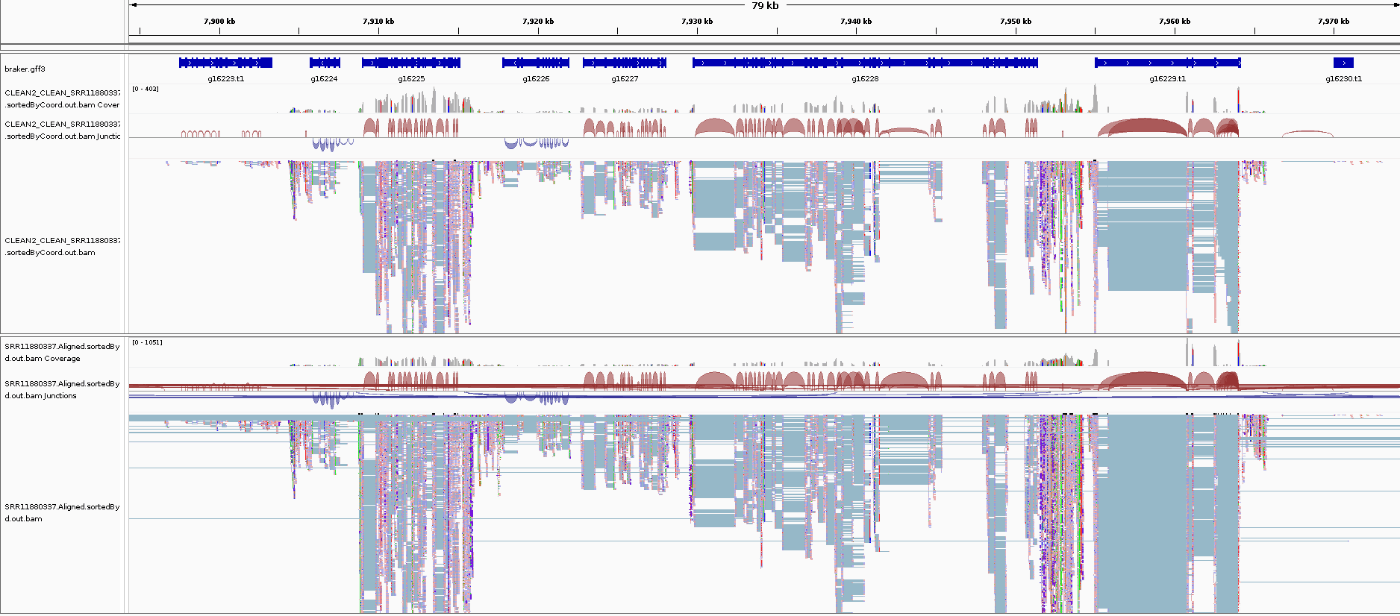
(上): TrinityとSTARで処理した後に対象種のゲノムにマッピング, (下):何も処理せず対象種のゲノムにマッピング
転写境界がある程度明瞭なマッピング結果になります。このデータ除去フローは以下の記事を参考にしています。
RNAseqの配列データの取得
以下の記事を参考に、fastqファイルを取得してfastpによるqcまで済ませておきましょう。
TrinityによるDe novo assembly
Trinity --seqType fq \
--left sra/SRR1533707_qc_1.fastq.gz,sra/SRR1533708_qc_1.fastq.gz,sra/SRR1533709_qc_1.fastq.gz,sra/SRR1533710_qc_1.fastq.gz,sra/SRR1533711_qc_1.fastq.gz,sra/SRR1533712_qc_1.fastq.gz,sra/SRR1533713_qc_1.fastq.gz,sra/SRR1533714_qc_1.fastq.gz,sra/SRR1533715_qc_1.fastq.gz,sra/SRR1533716_qc_1.fastq.gz \
--right sra/SRR1533707_qc_2.fastq.gz,sra/SRR1533708_qc_2.fastq.gz,sra/SRR1533709_qc_2.fastq.gz,sra/SRR1533710_qc_2.fastq.gz,sra/SRR1533711_qc_2.fastq.gz,sra/SRR1533712_qc_2.fastq.gz,sra/SRR1533713_qc_2.fastq.gz,sra/SRR1533714_qc_2.fastq.gz,sra/SRR1533715_qc_2.fastq.gz,sra/SRR1533716_qc_2.fastq.gz \
--SS_lib_type FR \
--min_contig_length 300 \
--CPU 32 \
--max_memory 100G \
--output trinity_output
-
--SS_lib_type:
strand-specificライブラリータイプであることを提示
Paired-End('RF' or 'FR'), Single-End('F' or 'R'). -
--min_contig_length:
アセンブルされる最小Contig長
カレントディレクトリにtrinity_output.Trinity.fastaが出力されていればDe novo assemblyは完了です。
STARによるマッピング
RNAseqのデータをTrinityで作成したcontig配列にマッピングします。
まずはSTARの実行に必要なIndexを作成します。RAMのエラーがでたらlimitGenomeGenerateRAMの値かgenomeSAsparseDの値を調整します。
ただ、それでもエラーが出るならtrinity_output.Trinity.fastaに含まれる短い配列を除去して対応します。
STAR \
--runThreadN 32 \
--runMode genomeGenerate \
--genomeDir GCA_036571765.1_ASM3657176v1_genomic_SM_index \
--genomeSAindexNbases 10 \
--limitGenomeGenerateRAM 100000000000 \
--genomeFastaFiles ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna
STAR \
--runThreadN 32 \
--runMode genomeGenerate \
--genomeDir trinity_output.Trinity_index \
--genomeSAindexNbases 10 \
--limitGenomeGenerateRAM 100000000000 \
--genomeSAsparseD 3 \
--genomeFastaFiles trinity_output.Trinity.fasta
genomeSAsparseDに指定する値で結果が若干変わります。可能であればデフォルト値で実行するほうが良いでしょう。
次はSTARでマッピングです。for loopで各SRRサンプルのデータを連続でマッピングしています。
eval _qc_fq_arrays=("$(ls sra/SRR*_qc_1.fastq.gz --quoting-style=shell)")
qc_fq_arrays=${_qc_fq_arrays[@]/_qc_1.fastq.gz/}
for qc_fq in ${qc_fq_arrays[@]};do
STAR \
--runThreadN 32 \
--genomeDir trinity_output.Trinity \
--readFilesIn ${qc_fq}_qc_1.fastq.gz ${qc_fq}_qc_2.fastq.gz \
--outFileNamePrefix bam/${qc_fq}. \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes All \
--readFilesCommand zcat
done
Samtools bam2fqによるfastq再生成
STARでマッピングされた配列をfastqとしてピックアップします。
eval _qc_fq_arrays=("$(ls sra/SRR*_qc_1.fastq.gz --quoting-style=shell)")
qc_fq_arrays=${_qc_fq_arrays[@]/_qc_1.fastq.gz/}
for qc_fq in ${qc_fq_arrays[@]};do
samtools bam2fq -n \
-1 sra/CLEAN_${qc_fq}_qc_1.fastq.gz \
-2 sra/CLEAN_${qc_fq}_qc_2.fastq.gz \
bam/CLEAN_${qc_fq}.Aligned.sortedByCoord.out.bam
done
fastq_pairによるmate pair readの抽出
samtools bam2fqで作成したfastqがペア構造を維持しておらず以降の処理でエラーが発生することがあったのでpair readのみを抽出します。
for qc_fq in ${qc_fq_arrays[@]}; do
# Decompress
pigz -d -p 12 sra/CLEAN_${qc_fq}_qc_1.fastq.gz sra/CLEAN_${qc_fq}_qc_2.fastq.gz
# Extract pair read
fastq_pair \
sra/CLEAN_${qc_fq}_qc_1.fastq \
sra/CLEAN_${qc_fq}_qc_2.fastq
# Compress
pigz -p 32 \
sra/CLEAN_${qc_fq}_qc_1.fastq \
sra/CLEAN_${qc_fq}_qc_2.fastq
done
これでクリーンなRNAseqの配列データを得ることができました。
再度ゲノム配列にマッピング
ここではIntronの許容サイズを指定できる--alignIntronMaxオプションを使います。
Intronのサイズは生物によってかなり異なるようです。
また、魚類の中でもゼブラフィッシュとその他の硬骨魚類では傾向が異なるなどあるようです。
数値を決めるのが難しいですが、1本目の論文を参考に33000bpをIntronの許容サイズとしました。
for qc_fq in ${qc_fq_arrays[@]}; do
STAR \
--runThreadN 32 \
--genomeDir GCA_036571765.1_ASM3657176v1_genomic_SM_index \
--readFilesIn sra/CLEAN_${qc_fq}_qc_1.fastq.gz sra/CLEAN_${qc_fq}_qc_2.fastq.gz \
--outFileNamePrefix bam/CLEAN_${qc_fq}. \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes All \
--readFilesCommand zcat
--alignIntronMax 33000 \
--outSAMstrandField intronMotif
done
これでクリーンなRNAseqの配列データを対象種のゲノム配列にマッピングしたBamファイルを得ることができました。
GeMoMaの実行
GeMoMaはコマンドとオプションがかなり多くhelpメッセージも見づらいので、公式ドキュメントとにらめっこしながら進めるといいです。
Extract RNA-seq evidence (ERE)
GeMoMa EREはGeMoMaにおいて、RNAseqのデータをエクソン-イントロン境界やスプライス部位の特定・補強のために実行します。
GeMoMa ERE USAGE
Parameters of tool "Extract RNA-seq Evidence" (ERE, version: 1.9):
s - Stranded (Defines whether the reads are stranded. In case of FR_FIRST_STRAND, the first read of a read pair or the only read in case of single-end data is assumed to be located on forward strand of the cDNA, i.e., reverse to the mRNA orientation. If you are using Illumina TruSeq you should use FR_FIRST_STRAND., range={FR_UNSTRANDED, FR_FIRST_STRAND, FR_SECOND_STRAND}, default = FR_UNSTRANDED) = FR_UNSTRANDED
The following parameter(s) can be used multiple times:
m - mapped reads file (BAM/SAM files containing the mapped reads, type = bam,sam) = null
v - ValidationStringency (Defines how strict to be when reading a SAM or BAM, beyond bare minimum validation., range={STRICT, LENIENT, SILENT}, default = LENIENT) = LENIENT
u - use secondary alignments (allows to filter flags in the SAM or BAM, default = true) = true
c - coverage (allows to output the coverage, default = true) = true
mmq - minimum mapping quality (reads with a mapping quality that is lower than this value will be ignored, valid range = [0, 255], default = 40) = 40
mc - minimum context (only introns that have evidence of at least one split read with a minimal M (=(mis)match) stretch in the cigar string larger than or equal to this value will be used, valid range = [1, 1000000], default = 1) = 1
maximumcoverage - maximum coverage (optional parameter to reduce the size of coverage output files, coverage higher than this value will be reported as this value, valid range = [1, 10000], OPTIONAL) = null
f - filter by intron mismatches (filter reads by the number of mismatches around splits, range={NO, YES}, default = NO) = NO
No parameters for selection "NO"
Parameters for selection "YES":
r - region around introns (test region of this size around introns/splits for mismatches to the genome, valid range = [0, 100], default = 10) = 10
n - number of mismatches (number of mismatches allowed in regions around introns/splits, valid range = [0, 100], default = 3) = 3
t - target genome (The target genome file (FASTA). Should be in IUPAC code, type = fasta,fas,fa,fna,fasta.gz,fas.gz,fa.gz,fna.gz) = null
e - evidence long splits (require introns to have at least this number of times the supporting reads as their length deviates from the mean split length, valid range = [0.0, 100.0], default = 0.0) = 0.0
mil - minimum intron length (introns shorter than the minimum length are discarded and considered as contiguous, valid range = [0, 1000], default = 0) = 0
repositioning - repositioning (due to limitations in BAM/SAM format huge chromosomes need to be split before mapping. This parameter allows to undo the split mapping to real chromosomes and coordinates. The repositioning file has 3 columns: split_chr_name, original_chr_name, offset_in_original_chr, type = tabular, OPTIONAL) = null
outdir - The output directory, defaults to the current working directory (.) = .
複数ファイルのままでは煩雑なので1ファイルにまとめます。
# Bam files array
eval BAM_array=("$(ls bam/CLEAN_*.Aligned.sortedByCoord.out.bam --quoting-style=shell)")
# Merge bam files
samtools merge -@ 32 -o bam/Paa_sra_truseq_Fstrand_rnaseq.bam ${BAM_array[@]}
使用しているライブラリ調整キットはIllumina TruSeq Stranded Total RNA kitで、こちらはFirst strandの情報を保持するキットのようなので、sにはFR_FIRST_STRANDを指定します。
GeMoMa ERE \
s=FR_FIRST_STRAND \
m=bam/Paa_sra_truseq_Fstrand_rnaseq.bam \
outdir=output_genome
CheckIntrons and DenoiseIntrons
Exon-Intron junctionの配列をチェックします。
GeMoMa CheckIntrons \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna.gz \
i=output_genome/introns.gff \
outdir=output_genome
怪しいIntronを除去します。
GeMoMa DenoiseIntrons \
i=output_genome/introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=output_genome
Extractor
GeMoMaの入力ファイル作成コマンドです。CDS領域を含むfastaファイルとgffからそれらの情報を抽出したテーブル形式のファイルを作成します。
出力ファイル
-
assignment.tabular: タブ区切りテーブルファイル -
cds-parts.fasta: タンパク質fasta
# Danio rerio
GeMoMa Extractor \
Ambiguity=AMBIGUOUS \
a=ref/Danio_rerio.GRCz11.113.chr.gff3.gz \
g=ref/Danio_rerio.GRCz11.dna_sm.primary_assembly.fa.gz \
outdir=ref_GRCz11
# Oryzias latipes
GeMoMa Extractor \
Ambiguity=AMBIGUOUS \
a=ref/Oryzias_latipes.ASM223467v1.113.chr.gff3.gz \
g=ref/Oryzias_latipes.ASM223467v1.dna_sm.toplevel.fa.gz \
outdir=ref_ASM223467v1
# Esox lucius
GeMoMa Extractor \
Ambiguity=AMBIGUOUS \
a=ref/Esox_lucius.fEsoLuc1.pri.113.chr.gff3.gz \
g=ref/Esox_lucius.fEsoLuc1.pri.dna_sm.toplevel.fa.gz \
outdir=ref_fEsoLuc1.pri
# Oncorhynchus mykiss
GeMoMa Extractor \
Ambiguity=AMBIGUOUS \
a=ref/Oncorhynchus_mykiss.USDA_OmykA_1.1.113.chr.gff3.gz \
g=ref/Oncorhynchus_mykiss.USDA_OmykA_1.1.dna_sm.toplevel.fa.gz \
outdir=ref_USDA_OmykA_1.1
# Salmo salar
GeMoMa Extractor \
Ambiguity=AMBIGUOUS \
a=ref/Salmo_salar.Ssal_v3.1.113.chr.gff3.gz \
g=ref/Salmo_salar.Ssal_v3.1.dna_sm.toplevel.fa.gz \
outdir=ref_Ssal_v3.1
MMseqs2 (Many-against-Many sequence searching) によるタンパク質検索
MMseqs2を使用してExtractorで得られたCDS領域の配列データに対して相同性検索を実施します。
mkdir mmseq_map mmseqs_tmp gemoma_output
mmseqs createdb \
ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
mmseq_map/GenomeDB \
-v 2
function run_mmseqs(){
local query_name=$1
echo "Processing ${query_name}"
# Create mmseqs db
mkdir mmseqs_query_$query_name
mmseqs createdb \
ref_$query_name/cds-parts.fasta \
mmseqs_query_$query_name/cdsDB \
-v 2
# 相同性検索
mmseqs search \
mmseqs_query_$query_name/cdsDB \
mmseq_map/GenomeDB \
mmseqs_query_$query_name/$query_name.out \
mmseqs_tmp/tmp_$query_name \
-e 100.0 --threads 32 -s 8.5 -a -v 2 \
--comp-bias-corr 0 \
--max-seqs 500 \
--mask 0 \
--orf-start-mode 1
# 結果の変換
mmseqs convertalis \
mmseqs_query_$query_name/cdsDB \
mmseq_map/GenomeDB \
mmseqs_query_$query_name/$query_name.out \
gemoma_output/search_$query_name.txt \
--threads 32 \
-v 2 \
--format-output "query,target,pident,alnlen,mismatch,gapopen,qstart,qend,tstart,tend,evalue,bits,empty,raw,nident,empty,empty,empty,qframe,tframe,qaln,taln,qlen,tlen"
}
# Running
run_mmseqs "GRCz11"
run_mmseqs "Ssal_v3.1"
run_mmseqs "USDA_OmykA_1.1"
run_mmseqs "ASM223467v1"
run_mmseqs "fEsoLuc1.pri"
GeMoMa
必須パラメーター
| Parameter | Comment |
|---|---|
| s |
mmseqs convertalisの検索結果ファイル |
| c | mmseqs/tblastnで取得したCDS領域のタンパク質配列 |
| a | mmseqs/tblastnで取得したCDS割当情報ファイル |
| t | 検索対象ゲノムFastaファイル |
複数回の指定が出来るパラメーター
| Parameter | Comment |
|---|---|
| i | RNA-seqから得られる可能性のあるイントロン(GFF)、type = gff,gff3 |
| sort | 検索結果の並び替え |
| Score | スコアリングに関する動作: Trust, ReScore, ReAlign |
| coverage | ストランド情報 |
| coverage_forward/_reverse | bedgraphファイルを指定する |
| outdir | 出力フォルダ |
# fEsoLuc1.pri
GeMoMa -Xmx100g GeMoMa \
s=gemoma_output/search_fEsoLuc1.pri.txt \
c=ref_fEsoLuc1.pri/cds-parts.fasta \
a=ref_fEsoLuc1.pri/assignment.tabular \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
i=output_genome/introns.gff \
sort=true \
Score=ReAlign \
coverage=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=20241120_gemoma_output_fEsoLuc1.pri
# ASM223467v1
GeMoMa -Xmx100g GeMoMa \
s=gemoma_output/search_ASM223467v1.txt \
c=ref_ASM223467v1/cds-parts.fasta \
a=ref_ASM223467v1/assignment.tabular \
sort=true \
Score=ReAlign \
coverage=STRANDED \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
i=output_genome/introns.gff \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=Gemoma_output_ASM223467v1
# Ssal_v3.1
GeMoMa -Xmx100g GeMoMa \
s=gemoma_output/search_Ssal_v3.1.txt \
c=ref_Ssal_v3.1/cds-parts.fasta \
a=ref_Ssal_v3.1/assignment.tabular \
sort=true \
Score=ReAlign \
coverage=STRANDED \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
i=output_genome/introns.gff \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=20241120_gemoma_output_Ssal_v3.1
# USDA_OmykA_1.1
GeMoMa -Xmx100g GeMoMa \
s=gemoma_output/search_USDA_OmykA_1.1.txt \
c=ref_USDA_OmykA_1.1/cds-parts.fasta \
a=ref_USDA_OmykA_1.1/assignment.tabular \
sort=true \
Score=ReAlign \
coverage=STRANDED \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
i=output_genome/introns.gff \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=20241120_gemoma_output_USDA_OmykA_1.1
# GRCz11
GeMoMa -Xmx100g GeMoMa \
s=gemoma_output/search_GRCz11.txt \
c=ref_GRCz11/cds-parts.fasta \
a=ref_GRCz11/assignment.tabular \
sort=true \
Score=ReAlign \
coverage=STRANDED \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
i=output_genome/introns.gff \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
outdir=Gemoma_output_GRCz11
GeMoMa Annotation Filter
GAFはさまざまなソースからの遺伝子予測を組み合わせてフィルタリングし、共通の遺伝子予測結果を生成します。
処理動作についてはドキュメントに以下の記載がありました。
- 冗長な予測結果の識別
- アノテーションの属性(attributes)情報に基づき、オプションで選択するなどした基準を適用してフィルタリング
- 重複する予測結果のクラスターを決定して、予測結果の並び替えや関連する予測結果を抽出
また、GeMoMaで作成した結果だけでなく、異なるツールによって推定された予測結果を統合することも可能なようです。
# ASM223467v1
GeMoMa -Xmx100g GAF \
g=Gemoma_output_ASM223467v1/predicted_annotation.gff \
outdir=Gemoma_output_ASM223467v1
# fEsoLuc1
GeMoMa GAF \
g=gemoma_output_fEsoLuc1.pri/predicted_annotation.gff \
outdir=gemoma_output_fEsoLuc1.pri
# Ssal_v3.1
GeMoMa GAF \
g=gemoma_output_Ssal_v3.1/predicted_annotation.gff \
outdir=gemoma_output_Ssal_v3.1
# USDA_OmykA_1.1
GeMoMa GAF \
g=gemoma_output_USDA_OmykA_1.1/predicted_annotation.gff \
outdir=gemoma_output_USDA_OmykA_1.1
# GRCz11
GeMoMa -Xmx100g GAF \
g=Gemoma_output_GRCz11/predicted_annotation.gff \
outdir=Gemoma_output_GRCz11
AnnotationFinalizer
GeMoMaが生成した遺伝子アノテーション結果を最適化するツールです。アノテーション付きのコーディング領域の配列のUTRを予測して、遺伝子と転写産物の情報を生成できます。
一方、UTR領域の予測は、RNAseqのゲノム汚染や重複遺伝子・近接する遺伝子、Unstranded RNA libraryによって悪影響を受ける可能性がある。
そのためマッピングしたリードの前処理にEREを使用することが推奨される。
# ASM223467v1
GeMoMa -Xmx100g AnnotationFinalizer \
g=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
a=Gemoma_output_ASM223467v1/filtered_predictions.gff \
u=YES \
i=output_genome/denoised_introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
rename=NO \
outdir=Gemoma_output_ASM223467v1
# fEsoLuc1
GeMoMa AnnotationFinalizer \
g=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
a=gemoma_output_fEsoLuc1.pri/filtered_predictions.gff \
u=YES \
i=output_genome/denoised_introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
rename=NO \
outdir=gemoma_output_fEsoLuc1.pri
# Ssal_v3.1
GeMoMa AnnotationFinalizer \
g=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
a=gemoma_output_Ssal_v3.1/filtered_predictions.gff \
u=YES \
i=output_genome/denoised_introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
rename=NO \
outdir=gemoma_output_Ssal_v3.1
# USDA_OmykA_1.1
GeMoMa AnnotationFinalizer \
g=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
a=gemoma_output_USDA_OmykA_1.1/filtered_predictions.gff \
u=YES \
i=output_genome/denoised_introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
rename=NO \
outdir=gemoma_output_USDA_OmykA_1.1
# GRCz11
GeMoMa -Xmx100g AnnotationFinalizer \
g=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
a=Gemoma_output_GRCz11/filtered_predictions.gff \
u=YES \
i=output_genome/denoised_introns.gff \
c=STRANDED \
coverage_forward=output_genome/coverage_forward.bedgraph \
coverage_reverse=output_genome/coverage_reverse.bedgraph \
rename=NO \
outdir=Gemoma_output_GRCz11
一括実行する場合
GeMoMaPipeline
GeMoMaPipelineはこれまで示してきたような処理を一括実行することができます。
- Extract RNA-seq evidence (ERE)
- DenoiseIntrons
- Extractor
- external search (tblastn or mmseqs)
- Gene Model Mapper (GeMoMa)
- GeMoMa Annotation Filter (GAF)
- AnnnotationFinalizer
うまく実行できれば全アノテーション結果が1ファイルに収まるのでGeMoMaPipelineでの実行がおすすめです。
個別実行でエラーチェックなどをしつつAnnotationFinalizerまでの実行目処がついたらGeMoMaPipelineという流れですね。
GeMoMa -Xmx100g \
GeMoMaPipeline \
threads=32 \
o=true \
GeMoMa.Score=ReAlign \
AnnotationFinalizer.r=NO \
AnnotationFinalizer.t=YES \
AnnotationFinalizer.u=YES \
t=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
s=own \
i=Oryzias_latipes \
a=ref/Oryzias_latipes.ASM223467v1.113.chr.gff3.gz \
g=ref/Oryzias_latipes.ASM223467v1.dna_sm.toplevel.fa.gz \
s=own \
i=Esox_lucius \
a=ref/Esox_lucius.fEsoLuc1.pri.113.chr.gff3.gz \
g=ref/Esox_lucius.fEsoLuc1.pri.dna_sm.toplevel.fa.gz \
s=own \
i=Oncorhynchus_mykiss \
a=ref/Oncorhynchus_mykiss.USDA_OmykA_1.1.113.chr.gff3.gz \
g=ref/Oncorhynchus_mykiss.USDA_OmykA_1.1.dna_sm.toplevel.fa.gz \
s=own \
i=Salmo_salar \
a=ref/Salmo_salar.Ssal_v3.1.113.chr.gff3.gz \
g=ref/Salmo_salar.Ssal_v3.1.dna_sm.toplevel.fa.gz \
s=own \
i=Danio_rerio \
a=ref/Danio_rerio.GRCz11.113.chr.gff3.gz \
g=ref/Danio_rerio.GRCz11.dna_sm.primary_assembly.fa.gz \
outdir=GeMoMaPipeline_Out \
tblastn=false \
r=MAPPED \
ERE.s=FR_FIRST_STRAND \
ERE.m=bam/Paa_sra_truseq_Fstrand_rnaseq.bam \
2>&1 | tee GeMoMa_$(date +%Y%m%d_%H%M%S).log
出力
GeMoMaPipelineで一括実行した前提で簡単に結果を確認します。
- protocol_GeMoMaPipeline.txt
Statistics:
Job WAITING RUNNING INTERRUPTED FAILED SUCCEEDED
---------------------------------------------------------
MmseqsCreateDB 0 0 0 0 1
EREAndFill 0 0 0 0 1
ExtractorAndSplit 0 0 0 0 5
Mmseqs 0 0 0 0 5
GeMoMa 0 0 0 0 160
Cat 0 0 0 0 5
GAF 0 0 0 0 1
AnnotationFinalizer 0 0 0 0 1
Extractor 0 0 0 0 1
SyntenyChecker 0 0 0 0 1
No errors detected.
Elapsed time: 15538 seconds (4h 18m 58s)
最下段でエラーなしでパイプラインが正常終了していることが確認できます。約4時間で終了したみたいですね。
- final_annotation.gff
- reference_gene_table.tabular
- unfiltered_predictions_from_species_0.gff
- unfiltered_predictions_from_species_1.gff
- unfiltered_predictions_from_species_2.gff
- unfiltered_predictions_from_species_3.gff
- unfiltered_predictions_from_species_4.gff
*species*.gffは近縁種ごとの統合前のgffファイルのようです。目的は複数種のGFFの情報を統合したfinal_annotation.gffです。
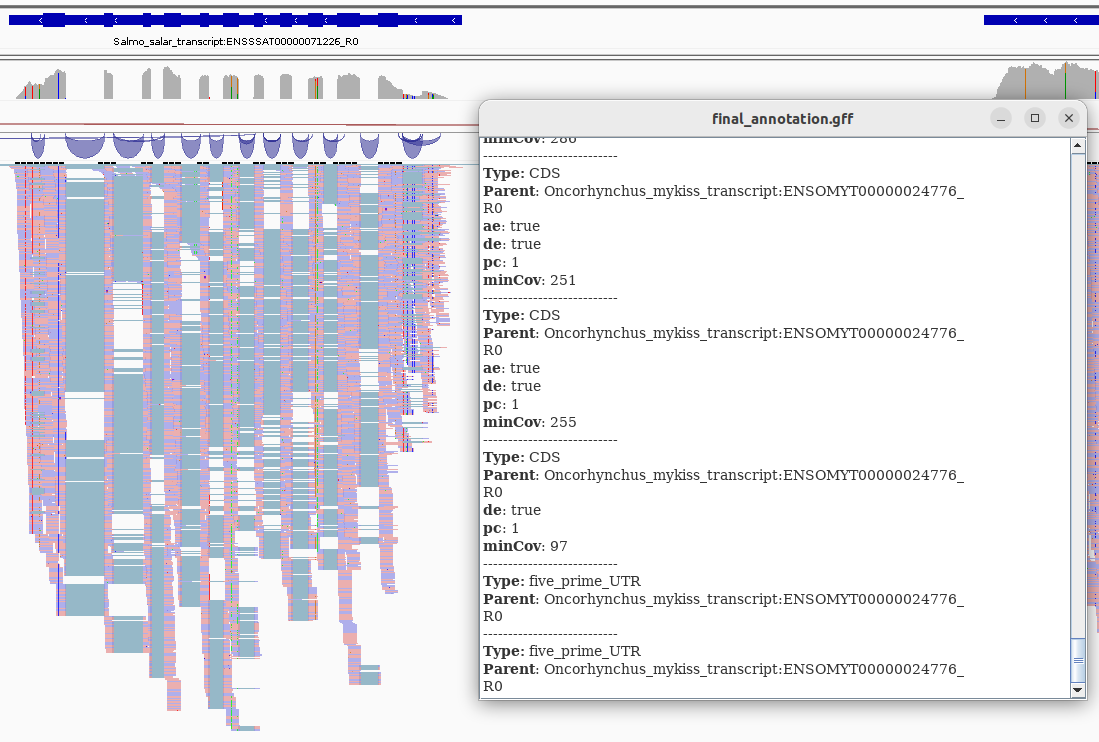
final_annotation.gffについて内容を確認すると、指定した種の遺伝子情報をもとにアユのリファレンスゲノムであるGCA_036571765.1_ASM3657176v1について遺伝子構造のアノテーションが付与できているのがわかります。
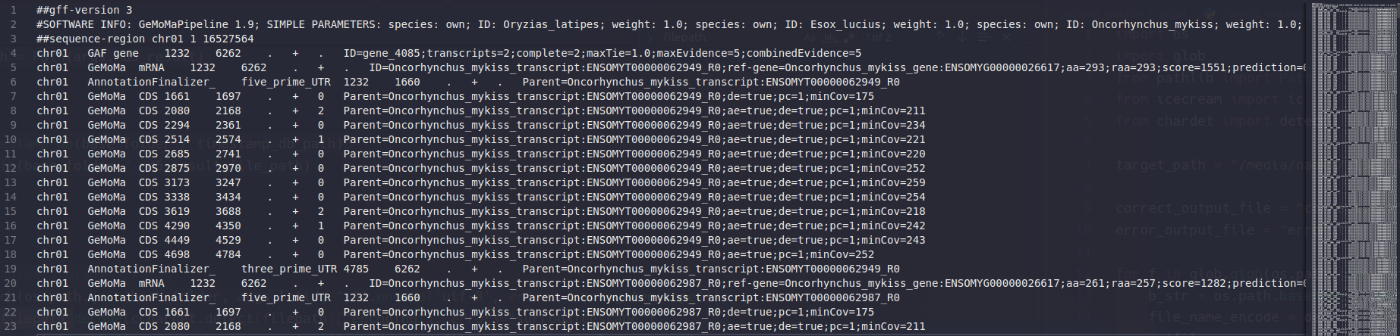
また、IGVで確認するとRNAseqの配列がマップされている領域でExon-Intron構造をいい感じに拾えているように見えます。
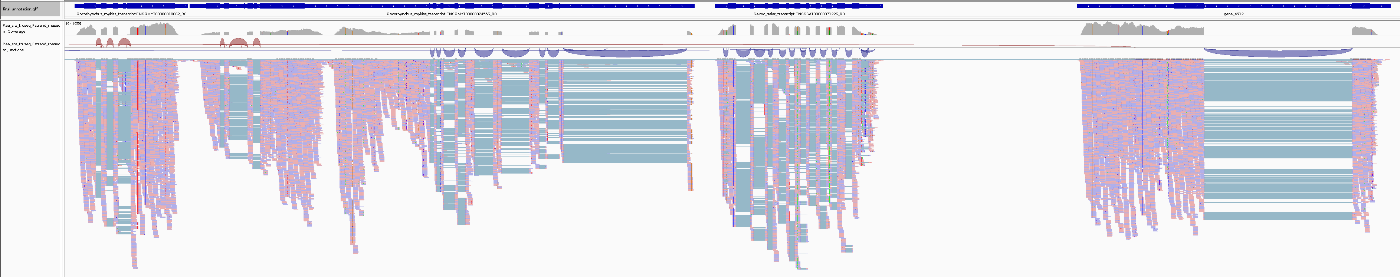
AnnotationFinalizer.u=YESを実行オプションに加ので3'UTRと5'UTRのアノテーションもGTFファイルに記載があります。ただ、現状では手探りなのでその妥当性はわかりません。
- predicted_proteins.fasta
遺伝子を予測した結果推定されたコーディング領域のプロテイン配列情報ファイル
終わり。
関連記事
参考
-
Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2203-5 -
genome_annotation_pipeline
https://github.com/KrabbenhoftLab/genome_annotation_pipeline -
Chromosome-level assembly of Cucumis sativus cv. ‘Tokiwa’ as a reference genome of Japanese cucumber
https://www.biorxiv.org/content/10.1101/2024.04.15.589484v2.full
Discussion