BRAKER3:遺伝子構造アノテーション

はじめに
Pool-seqやlcWGS(Low coverage whole genome resequencing)によるゲノムワイドな集団遺伝構造解析では、集団間で異なるSNP(一塩基多型)やSV(構造変異)の探索を行います。
集団間で異なる多型・変異マーカーの候補が得られた場合、どのような機能領域に生じた変異なのかの情報が欲しくなります。
上記に対する情報解析のアプローチとしては、ゲノムの構造や機能のアノテーションを実施するなどが挙げられます。
今回紹介するBRAKER3[1]は、ゲノムの構造のアノテーションに特化しているパイプラインです。
ここではそのBRAKER3を用いたゲノムの構造アノテーションについて説明して行きたいと思います。
ちなみに、BRAKERはリピート領域をマスク処理したゲノム配列が必要とします。以下の記事でRepeatModeler2[2]とRepeatMasker[3]を使用してアユのゲノム上のゲノム散在リピート配列にマスク処理を実施しているので、もしゲノム配列を新規に決定した場合には参考にしてください。
BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA
BRAKER3は、真核生物ゲノムの自動遺伝子予測パイプラインで、RNA-seqデータとタンパク質の相同性データを深層学習に利用します。
GeneMarkとAUGUSTUS[4]を組み合わせて解析を行う従来のBRAKER1[5]やBRAKER2[6]は、使用できるデータがRNA-seq or タンパク質データのいずれかに限られていました。
その後、GeneMark_ETP[7]とコンバイナーツールのTSEBRA[8]の開発により、データの統合が可能になりました。それにより、BRAKER3は以前より遺伝子予測の精度を向上させることができるようになりました。
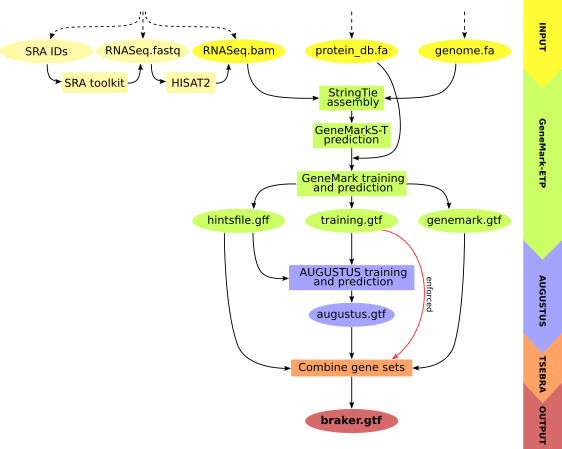
論文 Figure 1
BRAKER3のインストール
パイプライン化されているBRAKERはツールの依存関係が複雑なため、singularityやDockerといったコンテナサービスの利用が推奨されています。
今回は以下の条件を前提として進めます。
- root権限を持つユーザー (自身で所有しているPCなど)
- singularityがインストール済
- singularity v4.1.2のインストール例 (https://zenn.dev/edna_startup/articles/438d5d96342c29)
- WSL2 + UbuntuなどのLinux環境
dockerhubからBRAKER3のsingularityコンテナを作成します。
- dockerhub: https://hub.docker.com/r/teambraker/braker3
# 作業ディレクトリの作成
mkdir braker3 && braker3
# コンテナ用フォルダの作成
mkdir container && cd container
コンテナのBuild
singularity build --fakeroot braker3.0.8.sif docker://teambraker/braker3:latest
実行の練習も兼ねてバージョンを確認
singularity exec braker3.0.8.sif braker.pl --version
# braker.pl version 3.0.8
USAGE BRAKER v3.0.8
DESCRIPTION
braker.pl Pipeline for predicting genes with GeneMark-EX and AUGUSTUS with
RNA-Seq and/or proteins
SYNOPSIS
braker.pl [OPTIONS] --genome=genome.fa {--bam=rnaseq.bam | --prot_seq=prot.fa}
INPUT FILE OPTIONS
--genome=genome.fa fasta file with DNA sequences
--bam=rnaseq.bam bam file with spliced alignments from RNA-Seq
--prot_seq=prot.fa A protein sequence file in multi-fasta
format used to generate protein hints.
Unless otherwise specified, braker.pl will
run in "EP mode" or "ETP mode which uses
ProtHint to generate protein hints and
GeneMark-EP+ or GeneMark-ETP to
train AUGUSTUS.
--hints=hints.gff Alternatively to calling braker.pl with a
bam or protein fasta file, it is possible to
call it with a .gff file that contains
introns extracted from RNA-Seq and/or
protein hints (most frequently coming
from ProtHint). If you wish to use the
ProtHint hints, use its
"prothint_augustus.gff" output file.
This flag also allows the usage of hints
from additional extrinsic sources for gene
prediction with AUGUSTUS. To consider such
additional extrinsic information, you need
to use the flag --extrinsicCfgFiles to
specify parameters for all sources in the
hints file (including the source "E" for
intron hints from RNA-Seq).
In ETP mode, this option can be used together
with --geneMarkGtf and --traingenes to provide
BRAKER with results of a previous GeneMark-ETP
run, so that the GeneMark-ETP step can be
skipped. In this case, specify the hintsfile of
a previous BRAKER run here, or generate a
hintsfile from the GeneMark-ETP working
directory with the script get_etp_hints.py.
--rnaseq_sets_ids=SRR1111,SRR1115 IDs of RNA-Seq sets that are either in
one of the directories specified with
--rnaseq_sets_dir, or that can be downloaded
from SRA. If you want to use local files, you
can use unaligned reads in FASTQ format
(they have to be named ID.fastq if unpaired or
ID_1.fastq, ID_2.fastq if paired), or aligned reads
as a BAM file (named ID.bam).
--rnaseq_sets_dir=/path/to/rna_dir1 Locations where the local files of RNA-Seq data
reside that were specified with --rnaseq_sets_ids.
FREQUENTLY USED OPTIONS
--species=sname Species name. Existing species will not be
overwritten. Uses Sp_1 etc., if no species
is assigned
--AUGUSTUS_ab_initio output ab initio predictions by AUGUSTUS
in addition to predictions with hints by
AUGUSTUS
--softmasking_off Turn off softmasking option (enables by
default, discouraged to disable!)
--esmode Run GeneMark-ES (genome sequence only) and
train AUGUSTUS on long genes predicted by
GeneMark-ES. Final predictions are ab initio
--gff3 Output in GFF3 format (default is gtf
format)
--threads Specifies the maximum number of threads that
can be used during computation. Be aware:
optimize_augustus.pl will use max. 8
threads; augustus will use max. nContigs in
--genome=file threads.
--workingdir=/path/to/wd/ Set path to working directory. In the
working directory results and temporary
files are stored
--nice Execute all system calls within braker.pl
and its submodules with bash "nice"
(default nice value)
--alternatives-from-evidence=true Output alternative transcripts based on
explicit evidence from hints (default is
true).
--fungus GeneMark-EX option: run algorithm with
branch point model (most useful for fungal
genomes)
--crf Execute CRF training for AUGUSTUS;
resulting parameters are only kept for
final predictions if they show higher
accuracy than HMM parameters.
--keepCrf keep CRF parameters even if they are not
better than HMM parameters
--makehub Create track data hub with make_hub.py
for visualizing BRAKER results with the
UCSC GenomeBrowser
--busco_lineage=lineage If you provide a BUSCO lineage, BRAKER will
run compleasm on genome level to generate hints
from BUSCO to enhance BUSCO discovery in the
protein set. Also, if you provide a BUSCO
lineage, BRAKER will run compleasm to assess
the protein sets that go into TSEBRA combination,
and will determine the TSEBRA mode to maximize
BUSCO. Do not provide a busco_lineage if you
want to determina natural BUSCO sensivity of
BRAKER!
--email E-mail address for creating track data hub
--version Print version number of braker.pl
--help Print this help message
CONFIGURATION OPTIONS (TOOLS CALLED BY BRAKER)
--AUGUSTUS_CONFIG_PATH=/path/ Set path to config directory of AUGUSTUS
(if not specified as environment
variable). BRAKER1 will assume that the
directories ../bin and ../scripts of
AUGUSTUS are located relative to the
AUGUSTUS_CONFIG_PATH. If this is not the
case, please specify AUGUSTUS_BIN_PATH
(and AUGUSTUS_SCRIPTS_PATH if required).
The braker.pl commandline argument
--AUGUSTUS_CONFIG_PATH has higher priority
than the environment variable with the
same name.
--AUGUSTUS_BIN_PATH=/path/ Set path to the AUGUSTUS directory that
contains binaries, i.e. augustus and
etraining. This variable must only be set
if AUGUSTUS_CONFIG_PATH does not have
../bin and ../scripts of AUGUSTUS relative
to its location i.e. for global AUGUSTUS
installations. BRAKER1 will assume that
the directory ../scripts of AUGUSTUS is
located relative to the AUGUSTUS_BIN_PATH.
If this is not the case, please specify
--AUGUSTUS_SCRIPTS_PATH.
--AUGUSTUS_SCRIPTS_PATH=/path/ Set path to AUGUSTUS directory that
contains scripts, i.e. splitMfasta.pl.
This variable must only be set if
AUGUSTUS_CONFIG_PATH or AUGUSTUS_BIN_PATH
do not contains the ../scripts directory
of AUGUSTUS relative to their location,
i.e. for special cases of a global
AUGUSTUS installation.
--BAMTOOLS_PATH=/path/to/ Set path to bamtools (if not specified as
environment BAMTOOLS_PATH variable). Has
higher priority than the environment
variable.
--GENEMARK_PATH=/path/to/ Set path to GeneMark-ET (if not specified
as environment GENEMARK_PATH variable).
Has higher priority than environment
variable.
--SAMTOOLS_PATH=/path/to/ Optionally set path to samtools (if not
specified as environment SAMTOOLS_PATH
variable) to fix BAM files automatically,
if necessary. Has higher priority than
environment variable.
--PROTHINT_PATH=/path/to/ Set path to the directory with prothint.py.
(if not specified as PROTHINT_PATH
environment variable). Has higher priority
than environment variable.
--DIAMOND_PATH=/path/to/diamond Set path to diamond, this is an alternative
to NCIB blast; you only need to specify one
out of DIAMOND_PATH or BLAST_PATH, not both.
DIAMOND is a lot faster that BLAST and yields
highly similar results for BRAKER.
--BLAST_PATH=/path/to/blastall Set path to NCBI blastall and formatdb
executables if not specified as
environment variable. Has higher priority
than environment variable.
--COMPLEASM_PATH=/path/to/compleasm Set path to compleasm (if not specified as
environment variable). Has higher priority
than environment variable.
--PYTHON3_PATH=/path/to Set path to python3 executable (if not
specified as envirnonment variable and if
executable is not in your $PATH).
--JAVA_PATH=/path/to Set path to java executable (if not
specified as environment variable and if
executable is not in your $PATH), only
required with flags --UTR=on and --addUTR=on
--GUSHR_PATH=/path/to Set path to gushr.py exectuable (if not
specified as an environment variable and if
executable is not in your $PATH), only required
with the flags --UTR=on and --addUTR=on
--MAKEHUB_PATH=/path/to Set path to make_hub.py (if option --makehub
is used).
--CDBTOOLS_PATH=/path/to cdbfasta/cdbyank are required for running
fix_in_frame_stop_codon_genes.py. Usage of
that script can be skipped with option
'--skip_fixing_broken_genes'.
EXPERT OPTIONS
--augustus_args="--some_arg=bla" One or several command line arguments to
be passed to AUGUSTUS, if several
arguments are given, separate them by
whitespace, i.e.
"--first_arg=sth --second_arg=sth".
--skipGeneMark-ES Skip GeneMark-ES and use provided
GeneMark-ES output (e.g. provided with
--geneMarkGtf=genemark.gtf)
--skipGeneMark-ET Skip GeneMark-ET and use provided
GeneMark-ET output (e.g. provided with
--geneMarkGtf=genemark.gtf)
--skipGeneMark-EP Skip GeneMark-EP and use provided
GeneMark-EP output (e.g. provided with
--geneMarkGtf=genemark.gtf)
--skipGeneMark-ETP Skip GeneMark-ETP and use provided
GeneMark-ETP output (e.g. provided with
--gmetp_results_dir=GeneMark-ETP/)
--geneMarkGtf=file.gtf If skipGeneMark-ET is used, braker will by
default look in the working directory in
folder GeneMarkET for an already existing
gtf file. Instead, you may provide such a
file from another location. If geneMarkGtf
option is set, skipGeneMark-ES/ET/EP/ETP is
automatically also set. Note that gene and
transcript ids in the final output may not
match the ids in the input genemark.gtf
because BRAKER internally re-assigns these
ids.
In ETP mode, this option hast to be used together
with --traingenes and --hints to provide BRAKER
with results of a previous GeneMark-ETP run.
--gmetp_results_dir Location of results from a previous
GeneMark-ETP run, which will be used to
skip the GeneMark-ETP step. This option
can be used instead of --geneMarkGtf,
--traingenes, and --hints to skip GeneMark.
--rounds The number of optimization rounds used in
optimize_augustus.pl (default 5)
--skipAllTraining Skip GeneMark-EX (training and
prediction), skip AUGUSTUS training, only
runs AUGUSTUS with pre-trained and already
existing parameters (not recommended).
Hints from input are still generated.
This option automatically sets
--useexisting to true.
--useexisting Use the present config and parameter files
if they exist for 'species'; will overwrite
original parameters if BRAKER performs
an AUGUSTUS training.
--filterOutShort It may happen that a "good" training gene,
i.e. one that has intron support from
RNA-Seq in all introns predicted by
GeneMark-EX, is in fact too short. This flag
will discard such genes that have
supported introns and a neighboring
RNA-Seq supported intron upstream of the
start codon within the range of the
maximum CDS size of that gene and with a
multiplicity that is at least as high as
20% of the average intron multiplicity of
that gene.
--skipOptimize Skip optimize parameter step (not
recommended).
--skipIterativePrediction Skip iterative prediction in --epmode (does
not affect other modes, saves a bit of runtime)
--skipGetAnnoFromFasta Skip calling the python3 script
getAnnoFastaFromJoingenes.py from the
AUGUSTUS tool suite. This script requires
python3, biopython and re (regular
expressions) to be installed. It produces
coding sequence and protein FASTA files
from AUGUSTUS gene predictions and provides
information about genes with in-frame stop
codons. If you enable this flag, these files
will not be produced and python3 and
the required modules will not be necessary
for running brkaker.pl.
--skip_fixing_broken_genes If you do not have python3, you can choose
to skip the fixing of stop codon including
genes (not recommended).
--eval=reference.gtf Reference set to evaluate predictions
against (using evaluation scripts from GaTech)
--eval_pseudo=pseudo.gff3 File with pseudogenes that will be excluded
from accuracy evaluation (may be empty file)
--AUGUSTUS_hints_preds=s File with AUGUSTUS hints predictions; will
use this file as basis for UTR training;
only UTR training and prediction is
performed if this option is given.
--flanking_DNA=n Size of flanking region, must only be
specified if --AUGUSTUS_hints_preds is given
(for UTR training in a separate braker.pl
run that builds on top of an existing run)
--verbosity=n 0 -> run braker.pl quiet (no log)
1 -> only log warnings
2 -> also log configuration
3 -> log all major steps
4 -> very verbose, log also small steps
--downsampling_lambda=d The distribution of introns in training
gene structures generated by GeneMark-EX
has a huge weight on single-exon and
few-exon genes. Specifying the lambda
parameter of a poisson distribution will
make braker call a script for downsampling
of training gene structures according to
their number of introns distribution, i.e.
genes with none or few exons will be
downsampled, genes with many exons will be
kept. Default value is 2.
If you want to avoid downsampling, you have
to specify 0.
--checkSoftware Only check whether all required software
is installed, no execution of BRAKER
--nocleanup Skip deletion of all files that are typically not
used in an annotation project after
running braker.pl. (For tracking any
problems with a braker.pl run, you
might want to keep these files, therefore
nocleanup can be activated.)
DEVELOPMENT OPTIONS (PROBABLY STILL DYSFUNCTIONAL)
--splice_sites=patterns list of splice site patterns for UTR
prediction; default: GTAG, extend like this:
--splice_sites=GTAG,ATAC,...
this option only affects UTR training
example generation, not gene prediction
by AUGUSTUS
--overwrite Overwrite existing files (except for
species parameter files) Beware, currently
not implemented properly!
--extrinsicCfgFiles=file1,file2,... Depending on the mode in which braker.pl
is executed, it may require one ore several
extrinsicCfgFiles. Don't use this option
unless you know what you are doing!
--stranded=+,-,+,-,... If UTRs are trained, i.e.~strand-specific
bam-files are supplied and coverage
information is extracted for gene prediction,
create stranded ep hints. The order of
strand specifications must correspond to the
order of bam files. Possible values are
+, -, .
If stranded data is provided, ONLY coverage
data from the stranded data is used to
generate UTR examples! Coverage data from
unstranded data is used in the prediction
step, only.
The stranded label is applied to coverage
data, only. Intron hints are generated
from all libraries treated as "unstranded"
(because splice site filtering eliminates
intron hints from the wrong strand, anyway).
--optCfgFile=ppx.cfg Optional custom config file for AUGUSTUS
for running PPX (currently not
implemented)
--grass Switch this flag on if you are using braker.pl
for predicting genes in grasses with
GeneMark-EX. The flag will enable
GeneMark-EX to handle GC-heterogenicity
within genes more properly.
NOTHING IMPLEMENTED FOR GRASS YET!
--transmasked_fasta=file.fa Transmasked genome FASTA file for GeneMark-EX
(to be used instead of the regular genome
FASTA file).
--min_contig=INT Minimal contig length for GeneMark-EX, could
for example be set to 10000 if transmasked_fasta
option is used because transmasking might
introduce many very short contigs.
--translation_table=INT Change translation table from non-standard
to something else.
DOES NOT WORK YET BECAUSE BRAKER DOESNT
SWITCH TRANSLATION TABLE FOR GENEMARK-EX, YET!
--gc_probability=DECIMAL Probablity for donor splice site pattern GC
for gene prediction with GeneMark-EX,
default value is 0.001
--gm_max_intergenic=INT Adjust maximum allowed size of intergenic
regions in GeneMark-EX. If not used, the value
is automatically determined by GeneMark-EX.
--traingenes=file.gtf Training genes that are used instead of training
genes generated with GeneMark.
In ETP mode, this option can be used together
with --geneMarkGtf and --hints to provide BRAKER
with results of a previous GeneMark-ETP run, so
that the GeneMark-ETP step can be skipped.
In this case, use training.gtf from that run as
argument.
--UTR=on create UTR training examples from RNA-Seq
coverage data; requires options
--bam=rnaseq.bam.
Alternatively, if UTR parameters already
exist, training step will be skipped and
those pre-existing parameters are used.
DO NOT USE IN CONTAINER!
TRY NOT TO USE AT ALL!
--addUTR=on Adds UTRs from RNA-Seq coverage data to
augustus.hints.gtf file. Does not perform
training of AUGUSTUS or gene prediction with
AUGUSTUS and UTR parameters.
DO NOT USE IN CONTAINER!
TRY NOT TO USE AT ALL!
EXAMPLE
To run with RNA-Seq
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--bam=accepted_hits.bam
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--hints=rnaseq.gff
To run with protein sequences
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--prot_seq=proteins.fa
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--hints=prothint_augustus.gff
To run with RNA-Seq and protein sequences
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--prot_seq=proteins.fa --rnaseq_sets_ids=id_rnaseq1,id_rnaseq2 \
--rnaseq_sets_dir=/path/to/local/rnaseq/files
braker.pl [OPTIONS] --genome=genome.fa --species=speciesname \
--prot_seq=proteins.fa --bam=id_rnaseq1.bam,id_rnaseq2.bam
RNAseqのデータの準備
NCBI SRAに登録されているアユのRNAseqのデータを使用します。
以下のAccのものを使用することにしました。
| ACC | strand | bp |
|---|---|---|
| SRR11880337,SRR11880338 | 不明 | 100bp PE |
| SRR1533707 ~ SRR1533716 | Strand | 100bp PE |
| SRR17776511 ~ SRR17776525 | 不明 | 150 PE |
BRAKER3はFastq inputでも実行できるようですが、先にSTAR[9]でMappingします(BRAKERの推奨はHISAT2[10])。
HISAT2とSTARのどちらがいいのかについては以下の日本語記事が分かりやすかったです。
私は32スレッド,128GBのRAMが使用できるのでMapping結果が優れているSTARを選択しました。
LapTopではSTARの実行自体が厳しいかもしれませんが、BRAKERの実行もそこそこのスペックのPCでないと実行時間が膨れ上がります。なのでここで引っかかるようであれば解析環境を見直す必要があるかもしれません。
配列情報の準備とQC & Mapping
配列のダウンロードから、QC & Mapping作業の一部を示します。
ツールの準備
カレントディレクトリ下にenvフォルダを作成して、そこにcondaの仮想環境を作成します。
mkdir env && cd env
mamba create -p ${PWD}/braker_support_tools -c bioconda -c conda-forge -c -c mxmlnkn \
star=2.7.11b \
fastp=0.23.4 \
pigz=2.8 \
aria2=1.36 \
sra-tools=3.1.1 \
rapidgzip=0.12.0 \
git=2.46.2 \
samtools=1.21 \
seqkit=2.8.2
Activate
mamba activate ${PWD}/braker_support_tools
確認
fastp --version
# fastp 0.23.4
STAR --version
# 2.7.11b
pigz --version
# pigz 2.8
aria2c --version | grep "aria2 version"
# aria2 version 1.36.0
fasterq-dump --version
# fasterq-dump : 3.1.1
rapidgzip --help
# rapidgzip, CLI to the parallelized, indexed, and seekable gzip decoding library rapidgzip version 0.12.0
samtools --version
# samtools 1.21
# Using htslib 1.21
git --version
# git version 2.46.2
seqkit version
# seqkit v2.8.2
インストールしたツールを使用するときはactivateして使用する。sinfularityでのプログラム実行時はdeactivateして実行している。
配列情報のダウンロード
SRR1533707 ~ SRR1533716をダウンロードする例を示します。
mkdir sra && cd sra
for i in {07..16}; do
aria2c -x 15 -ctrue https://sra-pub-run-odp.s3.amazonaws.com/qc/SRR15337${i}/SRR15337${i}
done
SRR1533707 ~ SRR1533716という名称でSRAファイルが手に入ったのでfastqファイルに変換。
mkdir fastq
for i in {07..16}; do
fasterq-dump -p -x -S -e 32 sra/SRR15337${i} -O fastq
done
pigz -p 32 fastq/*.fastq
これで、fastqフォルダに*_1.fastq.gz,*_2.fastq.gz ファイルが作成されていればOKです。
配列情報のQC
fastpを用いてfastqファイルのqcを行います。こちらもSRR1533707 ~ SRR1533716を使った一例です。
mkdir qc
for i in {07..16}; do
fastp \
--in1 "sra/SRR15337${i}_1.fastq.gz" \
--in2 "sra/SRR15337${i}_2.fastq.gz" \
--out1 "qc/SRR15337${i}_qc_1.fastq.gz" \
--out2 "qc/SRR15337${i}_qc_2.fastq.gz" \
--html "qc/SRR15337${i}_qc_report.html" \
--json "qc/SRR15337${i}_qc_report.json" \
--qualified_quality_phred 30 \
--length_required 50 \
--detect_adapter_for_pe \
--trim_poly_g \
--cut_tail \
--thread 16
done
配列情報のMapping
Fastpを使ったQCが終了したらSTARを使ってゲノム配列にRNAをマッピングします。ゲノム配列は未マスキングかSoftmask処理したものが推奨されています。今回はSoftmask処理した配列を使用しました。
STARのindexを作成
NCBIのFTP siteに登録されているアユのゲノムをダウンロードします。
- Genome fasta: GCA_036571765.1_ASM3657176v1_genomic.fna.gz
mkdir ref && cd ref
# DL
aria2c -x 16 -ctrue https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/036/571/765/GCA_036571765.1_ASM3657176v1/GCA_036571765.1_ASM3657176v1_genomic.fna.gz
# Decompress
rapidgzip -d GCA_036571765.1_ASM3657176v1_genomic.fna.gz
BRAKERはheaderでエラーが生じやすいので単純なものに変更します。rename用の対応ファイルはこちらのものを使用しました。
- rename_GCA_036571765.1.tsv
seqkit replace -K -j 8 -p '(.+)$' -r '{kv}' -k rename_GCA_036571765.1.tsv GCA_036571765.1_ASM3657176v1_genomic.fna -o GCA_036571765.1_ASM3657176v1_genomic_SM.fna
# 一応半角スペースをinplaceで置換
sed -i "s/ /_/g" GCA_036571765.1_ASM3657176v1_genomic_SM.fna
chrかscaffoldの形式に置換できたのでSTARのindexファイルを作成します。
STAR \
--runThreadN 32 \
--runMode genomeGenerate \
--genomeDir ref/GCA_036571765.1_ASM3657176v1_genomic_SM_index \
--genomeSAindexNbases 13 \
--genomeFastaFiles ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna
Oct 05 15:18:18 ..... started STAR run
Oct 05 15:18:18 ... starting to generate Genome files
Oct 05 15:18:26 ... starting to sort Suffix Array. This may take a long time...
Oct 05 15:18:30 ... sorting Suffix Array chunks and saving them to disk...
Oct 05 15:19:13 ... loading chunks from disk, packing SA...
Oct 05 15:19:23 ... finished generating suffix array
Oct 05 15:19:23 ... generating Suffix Array index
Oct 05 15:19:58 ... completed Suffix Array index
Oct 05 15:19:58 ... writing Genome to disk ...
Oct 05 15:19:59 ... writing Suffix Array to disk ...
Oct 05 15:20:01 ... writing SAindex to disk
Oct 05 15:20:01 ..... finished successfully
finished successfullyと出ていればOKです。
STARによるMapping
こちらもSRR1533707 ~ SRR1533716を使った一例です。
mkdir bam
for i in {07..16}; do
STAR \
--runThreadN 32 \
--genomeDir ref/GCA_036571765.1_ASM3657176v1_genomic_SM_index \
--readFilesIn qc/SRR15337${i}_fq_1.fastq.gz qc/SRR15337${i}_fq_2.fastq.gz \
--outFileNamePrefix bam/SRR15337${i}. \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes All \
--readFilesCommand zcat
done
Oct 05 15:27:37 ..... started STAR run
Oct 05 15:27:37 ..... loading genome
Oct 05 15:27:39 ..... started mapping
Oct 05 15:28:51 ..... finished mapping
Oct 05 15:28:52 ..... started sorting BAM
Oct 05 15:29:09 ..... finished successfully
finished successfullyと出ていればOKです。1つのBamにまとめます。
# Bam files array
eval BAM_array=("$(ls bam/*.sortedByCoord.out.bam --quoting-style=shell)")
# Merge bam files
samtools merge -@ 32 -o bam/Paa_sra_rnaseq.bam ${BAM_array[@]}
タンパク質配列の準備
OrthoDBから対象種が属する分類階級に対応するfastaファイルをダウンロードします。
-
Metazoa.fa.gz
Size: 12GB -
Vertebrata.fa.gz
Size: 5.8GB -
Viridiplantae.fa.gz
Size: 3.0GB -
Arthropoda.fa.gz
Size: 4.4GB -
Fungi.fa.gz
Size: 7.2GB -
Alveolata.fa.gz
Size: 444MB -
Stramenopiles.fa.gz
Size: 343MB
Comment: BRAKERの場合は別の系統のタンパク質と組み合わせるか、Eukaryota.fa.gz を使用する必要あり -
Amoebozoa.fa.gz
Size: 48MB
Comment: BRAKERの場合は別の系統のタンパク質と組み合わせるか、Eukaryota.fa.gz を使用する必要あり -
Euglenozoa.fa.gz
Size: 106MB
Comment: BRAKERの場合は別の系統のタンパク質と組み合わせるか、Eukaryota.fa.gz を使用する必要あり
上記のいずれのニーズも満たさない場合には、**Eukaryota.fa.gz(23GB)**を使用する。
アユは脊椎動物なのでVertebrata.fa.gzをダウンロードします。ダウンロードスピードがめちゃくちゃ遅く、150KB/s 程度しかでないので気長に待つ(8時間くらい)。
OrthoDB v12のデータをDLした時は許容できるスピードでした。
https://bioinf.uni-greifswald.de/bioinf/partitioned_odb11/
# 移動
cd ref
# Download
wget https://bioinf.uni-greifswald.de/bioinf/partitioned_odb12/Vertebrata.fa.gz
解凍していないとheader名のエラーが出るらしいので解凍しておきます。
rapidgzip -d Vertebrata.fa.gz
BRAKER3による遺伝子構造アノテーション
まずはAugustusのconfigの設定をします。
基本的にホームディレクトリ下の.augustus内にconfigフォルダが作成されてそこに必要なファイルなどが配置される仕様になっています。
ただ、今回はプロジェクト単位の運用のことも考えてSingularityコンテナを使用していますが、Singularityコンテナはパイプランに実行中にコンテナ内部にファイルを作成することが多分できません。
そうなると自動でconfigファイルが作成されずエラーが発生します。それを避けるためにカレントディレクトリ下にConfigファイルを作成してそこを指定します。使用するAugustusのバージョンにあったconfigファイル群を準備する作業を以下に示します。
# config配置用フォルダの作成と移動
mkdir -p config/AUGUSTUS_CONFIG_PATH && cd config/AUGUSTUS_CONFIG_PATH
# Augustus v3.5.0のリポジトリをクローン
git clone https://github.com/Gaius-Augustus/Augustus.git -b v3.5.0
# configフォルダをAUGUSTUS_CONFIG_PATHに移動
mv Augustus/config ./
# 不要なフォルダを削除
rm -rfv Augustus
braker.plの実行時に--AUGUSTUS_CONFIG_PATHでAugustusのconfigファイルの場所を指定します。
# Singularity setting
export SINGULARITY_BIND="$PWD:$PWD"
BRAKERの実行
singularity exec container/braker3.0.8.sif braker.pl \
--AUGUSTUS_CONFIG_PATH=config/AUGUSTUS_CONFIG_PATH/config \
--threads=32 \
--genome=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
--prot_seq=ref/Vertebrata.fa \
--species="Plecoglossus_altivelis" \
--workingdir=braker3_out_$(date +%y%m%d%H%M) \
--gff3 \
--busco_lineage="eukaryota_odb10" \
--verbosity=4 \
--bam=bam/Paa_sra_rnaseq.bam
指定しているパラメーター
-
--AUGUSTUS_CONFIG_PATH: AUGUSTUSの設定ディレクトリへのパスを指定(環境変数として指定されていない場合) -
--threads: 計算に使用する最大スレッド数を指定。注意:optimize_augustus.plは最大8スレッドを使用し、augustusは指定したゲノム内のコンティグ数までスレッドを使用 -
--genome: BRAKERで解析するFASTA形式のDNA配列ファイルを指定 -
--prot_seq: FASTA形式のタンパク質配列ファイルを指定。BRAKERはこのファイルを使用して、タンパク質ヒントファイルを生成 -
--species: 対象種の名前を指定。既存の種は上書きされず、種名を指定しない場合、自動的に「Sp_1」などの名前が割り振られる -
--workingdir: 作業ディレクトリのパスを指定。このディレクトリに結果とtmpファイルが保存される -
--gff3: 出力をGFF3形式に指定(デフォルトはGTF形式) -
--busco_lineage: BUSCO系統を指定。指定すると、BRAKERはBUSCOを用いてゲノムレベルのヒントを生成し、BUSCO検出を強化。また、TSEBRAの組み合わせに含まれるタンパク質セットを評価し、BUSCOスコアを最大化するためのTSEBRAモードを決定 -
--verbosity: ログの詳細度を指定。0はログなし実行、1は警告のみログ、2は設定もログ、3はすべての主要ステップをログ、4は詳細なログを出力 -
--bam: RNA-Seqのスプライスアライメントを含むBAMファイルを指定。RNA-Seqデータからイントロンヒントを生成するために使用
BRAKER3の出力ファイル
出力ファイルの内いくつか後続解析に使用しそうなものを明記します。
braker.gtf
BRAKER3が出力する最終遺伝子定義ファイル。実行パターンによって異なる情報が記載されている
chr01 AUGUSTUS gene 1 859 . + . g1
chr01 AUGUSTUS transcript 1 859 1 + . g1.t1
chr01 AUGUSTUS CDS 1 145 1 + 0 transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS exon 1 145 . + . transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS intron 146 332 1 + . transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS CDS 333 583 1 + 2 transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS exon 333 583 . + . transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS intron 584 775 1 + . transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS CDS 776 859 1 + 0 transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS exon 776 859 . + . transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS stop_codon 857 859 . + 0 transcript_id "g1.t1"; gene_id "g1";
chr01 AUGUSTUS gene 1661 4784 . + . g2
chr01 AUGUSTUS transcript 1661 4784 0.85 + . g2.t1
chr01 AUGUSTUS start_codon 1661 1663 . + 0 transcript_id "g2.t1"; gene_id "g2";
...
-
ETPモード (今回実行): AUGUSTUS と GeneMark-ETP によって予測され、TSEBRA によって結合およびフィルタリングされた遺伝子で構成される BRAKER の最終遺伝子セット
-
ETPモード以外 : augustus.hints.gtfと信頼性の高いGeneMark-ES/ET/EP予測 (外部証拠によってサポートされている遺伝子情報) を統合。
-
--esmodeは、 augustus.ab_initio.gtfとGeneMark-ESを統合。感度は高いが擬陽性も増える可能性あり。
-
braker.codingseq
FASTA形式のコーディング領域の配列ファイル
>g1.t1
CCTGTGTTGTTTGGCTGTTCCATTTGGGAGAATATTCGCTTTGGGAAGCCTGAGGCAACC
GATGCTGAGGTCGTCAACGCTGCTAAGCAAGCCAACGCTCATCGCTTCATCACAGGCTTT
CCAGAGGGATACAACACTGTGGTCGGTGAACGTGGGGCAACTCTGTCAGGGGGGCAGAAG
CAGCGGATTGCCATTGCTCGTGCCCTGATCAAGAACCCCAGCATCCTGGTGTTGGATGAG
GCCACCAGTGCCCTTGATGCGGAGTCAGAGCGTGTGGTACAGGAGGCTTTGGACCGGGCC
ACTACAGGCCGGACCGTACTCATCATTGCGCATCGCCTCAGTACCATCCAGGGAGCTGAC
CTAATCTGTGTCATGAGCAACGGACGCATTGTAGAGGCAGGAACTCACTTGGACTTGCTG
AGCAAAGGAGGACTCTATGCGGAACTGATTCGTAGACaaagagcagaggaaaacaaCTGA
>g2.t1
ATGCAGAAATATGAAAAGCTCGAGAAAATCGGAGAAGGTACATATGGCACTGTTTTCAAA
GCTAAAAACCGAGAGACACATGAAATTGTAGCTTTAAAAAGAGTTAGACTGGATGACGAC
GACGAGGGAGTTCCTAGTTCTGCCTTGCGAGAGATTTGCCTTTTGAAAGAGCTGAAACAC
...
>g3.t1
ATGGTAAATGCACCCATGCCAAAACCAATCAGTCAGGGGTCTACAACACCCGACGATGCC
CTGGAGACTTCTGGAGGAGGCGCAGACAGTAATGAGCTGGAGCCCACATGGATACAGATT
ACAGTGGCTTTCATGAGAAGGACCAAAATCCACGGGCTGAAGTTTGTCTTCTCACCAGAC
AAGACCAGACTACAGCGGTTCCTCTGGCTCATTGCTTTCTTCATCTGTGTTGGACTGCTA
GCCACCTGGTCATGGAACAGGATCTTTTACTTGATGTCCTACCCGGCCGTCACGAAGATC
TACATGGTCTGGTCTCATAACATGTCCTTTCCAGCTGTTACTTTTTGTAACAAAAATGTA
braker.aa
FASTA形式のタンパク質配列ファイル
>g1.t1
PVLFGCSIWENIRFGKPEATDAEVVNAAKQANAHRFITGFPEGYNTVVGERGATLSGGQK
QRIAIARALIKNPSILVLDEATSALDAESERVVQEALDRATTGRTVLIIAHRLSTIQGAD
LICVMSNGRIVEAGTHLDLLSKGGLYAELIRRQRAEENN*
>g2.t1
MQKYEKLEKIGEGTYGTVFKAKNRETHEIVALKRVRLDDDDEGVPSSALREICLLKELKH
KNIVRLHDVLHSDKKLTLVFEYCDQDLKKYFDSCNGDLDPETVKSFMYQLLKGLAFCHSR
NVLHRDLKPQNLLINRNGELKLADFGLARAFGIPVRCYSAEVVTLWYRPPDVLFGAKLYS
TSIDMWSAGCIFAELANAGRPLFPGNDVDDQLKRIFRLLGTPTEEQWQTMTKLPDYKPYP
MYPATTSLVNVVPKLSSTGRDLLQNLLKCNPVQRISAEEALQHPYFADFCPP*
>g3.t1
MVNAPMPKPISQGSTTPDDALETSGGGADSNELEPTWIQITVAFMRRTKIHGLKFVFSPD
KTRLQRFLWLIAFFICVGLLATWSWNRIFYLMSYPAVTKIYMVWSHNMSFPAVTFCNKNV
FRVSSLTKADLYQSGYWMDLMYANHTVMESSLAILRDNHKHSLLSLLDFKDYSPPPHENV
DTTEMIGRLGHQLEDMLLECRFRGENCTHRNFSTIYTRYGKCYTFNSGLDGNPLLTTLKG
GTGNGLEIMLDIQQDEYLPVWGETDETSYEAGIKVQIHSQDEPPFIDQLGFGVAPGFQTF
VSCQQQLLQYLPPPWGDCKSTPIDSEFFSTYSITACRIDCETRYLVENCNCRMVHMPGSS
TVCTPEQYKDCADPALDFLVEKDNDYCVCETPCNMTRYGKELSMVKIPSKASAKYLAKKF
NKTEQYIGENILVMDIFFEALNYEKIEQKKAYEIAGLLGDIGGQMGLFIGASVLTILEIF
DYLYEVFKDKVFGYFLNKKRPRRCQSDNLEFPENPTRPGVTPNHAPRAPVTPSGVTRTVS
DTRRTCYLVTRL*
braker.gff3
gff3形式の最終遺伝子定義ファイル。--gff3が引数に指定された場合にのみ生成
chr01 AUGUSTUS gene 1 859 . + . ID=g1;
chr01 AUGUSTUS mRNA 1 859 1 + . ID=g1.t1;Parent=g1;
chr01 AUGUSTUS CDS 1 145 1 + 0 ID=g1.t1.CDS1;Parent=g1.t1;
chr01 AUGUSTUS exon 1 145 . + . ID=g1.t1.exon1;Parent=g1.t1;
chr01 AUGUSTUS intron 146 332 1 + . ID=g1.t1.intron1;Parent=g1.t1;
chr01 AUGUSTUS CDS 333 583 1 + 2 ID=g1.t1.CDS2;Parent=g1.t1;
chr01 AUGUSTUS exon 333 583 . + . ID=g1.t1.exon2;Parent=g1.t1;
chr01 AUGUSTUS intron 584 775 1 + . ID=g1.t1.intron2;Parent=g1.t1;
chr01 AUGUSTUS CDS 776 859 1 + 0 ID=g1.t1.CDS3;Parent=g1.t1;
chr01 AUGUSTUS exon 776 859 . + . ID=g1.t1.exon3;Parent=g1.t1;
chr01 AUGUSTUS stop_codon 857 859 . + 0 ID=g1.t1.stop1;Parent=g1.t1;
chr01 AUGUSTUS gene 1661 4784 . + . ID=g2;
chr01 AUGUSTUS mRNA 1661 4784 0.85 + . ID=g2.t1;Parent=g2;
chr01 AUGUSTUS start_codon 1661 1663 . + 0 ID=g2.t1.start1;Parent=g2.t1;
chr01 AUGUSTUS CDS 1661 1697 1 + 0 ID=g2.t1.CDS1;Parent=g2.t1;
chr01 AUGUSTUS exon 1661 1697 . + . ID=g2.t1.exon1;Parent=g2.t1;
chr01 AUGUSTUS intron 1698 2079 1 + . ID=g2.t1.intron1;Parent=g2.t1;
Augustusフォルダ内のファイル
Augustusの遺伝子定義ファイルやタンパク、塩基配列ファイル
- Augustus/augustus.hints.aa
- Augustus/augustus.hints.codingseq
- Augustus/augustus.hints.gff3
- Augustus/augustus.hints.gtf
GeneMark-E*/genemark.gtf
GeneMark-ES/ET/EP/EP+/ETPによって予測された遺伝子情報が記載された遺伝子定義ファイル
chr01 gmst stop_codon 5948245 5948247 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5948245 5948285 76.265959 - 2 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Terminal"; class "LORF_UPSTOP";
chr01 gmst intron 5948286 5948405 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5948406 5948499 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Internal"; class "LORF_UPSTOP";
chr01 gmst intron 5948500 5948617 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5948618 5948766 76.265959 - 2 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Internal"; class "LORF_UPSTOP";
chr01 gmst intron 5948767 5949006 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5949007 5949185 76.265959 - 1 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Internal"; class "LORF_UPSTOP";
chr01 gmst intron 5949186 5949546 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5949547 5949664 76.265959 - 2 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Internal"; class "LORF_UPSTOP";
chr01 gmst intron 5949665 5949767 76.265959 - 0 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; class "LORF_UPSTOP";
chr01 gmst CDS 5949768 5949931 76.265959 - 1 gene_id "MSTRG.252"; transcript_id "MSTRG.252.1"; status "complete"; cds_type "Internal"; class "LORF_UPSTOP";
hintsfile.gff
RNAseq.bamおよび/またはタンパク質データから抽出された外部証拠データ。
chr01 b2h intron 146 332 10 + . mult=10;pri=4;src=E
chr01 ProtHint intron 146 332 50 + . mult=50;src=M;pri=4
chr01 ProtHint intron 146 332 50 + . mult=50;src=P;pri=4;al_score=0.471865;
chr01 ProtHint intron 584 775 50 + . mult=50;src=P;pri=4;al_score=0.439666;
chr01 ProtHint intron 584 775 50 + . mult=50;src=M;pri=4
chr01 b2h intron 584 775 20 + . mult=20;pri=4;src=E
chr01 b2h intron 584 779 4 + . mult=4;pri=4;src=E
chr01 ProtHint stop 857 859 50 + 0 mult=50;src=P;pri=4;al_score=0.28595;
chr01 ProtHint stop 857 859 50 + 0 mult=50;src=M;pri=4
chr01 ProtHint start 1661 1663 2 + 0 src=P;mult=25;pri=4;al_score=0.469421;
braker_originalフォルダ
compleasm[11] を使用してBUSCOの完全性を向上させる前にBRAKER (TSEBRA) によって予測された遺伝子定義ファイルや配列データ
- braker_original/braker.aa
- braker_original/braker.codingseq
- braker_original/braker.gtf
bbcフォルダ
BRAKERの最終出力におけるBUSCOの完全性を向上させるために使用されるTSEBRAのbest_by_compleasm.pyスクリプトの出力フォルダ
- augustus/
- augustus_genemark_keep.gtf
- augustus_keep.gtf
- better/
- braker/
- genemark/
- genemark.aa
- genemark.codingseq
- genemark_keep.gtf
トラブルシュート
途中から記載し始めたので序盤のエラーは記載されていない or 文中にIssueとともに対処後の実行方法が記載されているが備忘録として。
etrainingのエラー
etrainingのエラー
ERROR in file /opt/BRAKER/scripts/braker.pl at line 6291
Failed to execute: /opt/Augustus/bin//etraining --species=Paa --AUGUSTUS_CONFIG_PATH=/home/naoki/.augustus <path to current dir>/braker3/braker3_out/train.f.gb 1> <path to current dir>/braker3/braker3_out/gbFilterEtraining.stdout 2><path to current dir>/braker3/braker3_out/errors/gbFilterEtraining.stderr
gbFilterEtraining.stdoutを見てみるとCould not locate command line parameters file: /home/<user name>/.augustus/parameters/aug_cmdln_parameters.json.との記載がある。
初期実行時にはAUGUSTUS_CONFIG_PATHを設定していなかったことと、過去に実行したときの古いフォルダが残っていたため発生したと考えられる。
Augustus and/or etraining within BRAKER complain that the file aug_cmdln_parameters.json is missing. Even though I am using the latest Singularity container!
BRAKER copies the AUGUSTUS_CONFIG_PATH folder to a writable location. In older versions of Augustus, that file was indeed not existing. If the local writable copy of a folder already exists, BRAKER will not re-copy it. Simply delete the old folder. (It is often ~/.augustus, so you can simply do rm -rf ~/.augustus; the folder might be residing in $PWD if your home directory was not writable).
rm -rf -rf ~/.augustusで消してしまえば、再度コピーされるらしい。ただ、バージョンに依存性があり上書きされないのであればホームディレクトリ下に置いての運用は面倒なので、AUGUSTUS_CONFIG_PATHをカレントディレクトリ下の特定フォルダに設定して再実行すれば問題ないはず。
今回はカレントディレクトリ下の特定フォルダを指定して実行した。
BRAKERのリスタート
ここに記載されている方法を参照する。
BRAKERのリスタート
いくつかリスタートポイントがあるようで、私の場合GeneMark-ETPが終了した後のAUGUSTUSのトレーニングから再実行するオプション2によるリスタートが必要でした。
まず、エラーが発生したBRAKERの結果フォルダを指定。
BRAKER_OLD=<path to old braker output dir>
GeneMarkETPの結果を手動でBRAKERに与える
-
--geneMarkGtf: GeneMarkの結果ファイルであるgenemark.gtfファイルのパス(ex. ${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf) -
--traingenes: GeneMarkで生成されたトレーニングファイル (ex. ${BRAKER_OLD}/GeneMark-ETP/training.gtf) -
--hints: RNA-Seqから抽出したイントロンやタンパク質のヒントを含む.gffファイル (ex. ${BRAKER_OLD}/hintsfile.gff)
リスタートは以下のような感じ。この時点のリスタートの理由はAUGUSTUS_CONFIG_PATHがホームディレクトリにある.augstusディレクトリを参照しており、バージョンアップでフォルダ構造が変わっていたことでエラーで止まっていたため。
Augustus v3.5.0のGitHubをcloneしてconfigフォルダのみを残す。
AUGUSTUS_CONFIG_PATH=$PWD/config/AUGUSTUS_CONFIG_PATH
cd $PWD/config/AUGUSTUS_CONFIG_PATH
git clone https://github.com/Gaius-Augustus/Augustus.git
mv Augustus/config ./
rm -rf Augustus/
singularity exec container/braker3.0.8.sif braker.pl \
--threads=32 \
--genome=ref/GCA_036571765.1_ASM3657176v1_genomic_SM.fna \
--species="Paa" \
--workingdir=braker3_out_$(date +%y%m%d%H%M) \
--gff3 \
--busco_lineage="eukaryota_odb10" \
--verbosity=4 \
--bam=bam/SRR11880337.Aligned.sortedByCoord.out.bam \
--geneMarkGtf=${BRAKER_OLD}/GeneMark-ETP/proteins.fa/genemark.gtf \
--traingenes=${BRAKER_OLD}/GeneMark-ETP/training.gtf \
--hints=${BRAKER_OLD}/hintsfile.gff \
--skipGeneMark-ETP \
--AUGUSTUS_CONFIG_PATH=${AUGUSTUS_CONFIG_PATH}/config
--prot_seqオプションは--hinsオプションと同時に使えないので削除してあります
上記コマンドはGeneMark−ETPを飛ばす形でcompleasmからリスタートされます。
おわり。
参考
- BRAKER3による遺伝子予測(ベンチマーク編)
https://hashiyuki.hatenablog.com/entry/2023/04/11/124655 - STAR-RSEMによる発現量推定 その2 (2020/08/20 追記)
https://rnakato.hatenablog.jp/entry/2018/12/28/184301 - Whole-Genome Annotation with BRAKER
https://math-inf.uni-greifswald.de/storages/uni-greifswald/fakultaet/mnf/mathinf/stanke/BRAKER-manuscript.pdf - Structural-Annotation
https://github.com/CBC-UCONN/Structural-Annotation - Galba: genome annotation with miniprot and AUGUSTUS
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05449-z
-
https://academic.oup.com/nar/article/34/suppl_2/W435/2505582 ↩︎
-
https://academic.oup.com/bioinformatics/article/32/5/767/1744611 ↩︎
-
https://academic.oup.com/nargab/article/3/1/lqaa108/6066535 ↩︎
-
https://academic.oup.com/bioinformatics/article/29/1/15/272537?login=false ↩︎
-
https://academic.oup.com/bioinformatics/article/39/10/btad595/7284108 ↩︎
Discussion
2024年12月時点では、Breaker3に用いるRNA-Seqのアラインメントデータの生成にSTARを用いる場合、
--outSAMstrandField intronMotifオプションを追加することが推奨されているようですコメント頂きありがとうございます。BrakerのGithubリポジトリのBRAKER with RNA-Seq dataの章に記載があることを確認しました。
--outSAMstrandField intronMotifつけてエラーがでないことを確認した後に更新したいと思います。