Tiberius:HMMを使用した遺伝子予測のためのEnd-to-Endの深層学習法

Tiberius: End-to-End Deep Learning with an HMM for Gene Prediction
遺伝子予測のためのHMMを用いたエンドツーエンドのディープラーニング
はじめに
遺伝子構造予測は、ゲノム内の遺伝子とそのExon-Intron構造を特定する解析であり、得られる結果は下流解析の基礎となります。
比較ゲノム解析で表現形質の違いを明らかにしたい場合、高精度のアノテーションが求められるが、現在の真核遺伝子予測の精度は十分と言えません。既存の予測パイプラインの多くは、過去に同定されたタンパク質の情報に加え、RNAseqのデータを統合して精度向上を図っています。しかし、RNAseqが利用できな種も多く、例えば哺乳類では全体の48%に相当する341種にRNAseqのデータがありません。RNAseqなしで高品質なゲノムアノテーションを実現できれば、大規模な時間とリソースの節約が可能です。
遺伝子構造予測は主に以下の2種類に大別されます:
- ab initio予測: ターゲットゲノムの情報のみを利用
- de novo予測: 複数のゲノム間の進化的情報も利用
ab initio予測は選択的スプライシングや重複遺伝子、ネスト遺伝子に対応できない点に注意が必要です。長らく使用されている隠れマルコフモデル ( HMM )は、多くのツールで採用されており、例としてNCBIのGeScanやBRAKERに内包されるAUGUSTUS, GeneMarkがあります。これらのツールはマルコフ連鎖やスプライスサイトの識別に関連する機械学習や統計技術を活用していますが、特定の系統群に対しては新たにトレーニングが必要です。
近年では、深層学習を取り得れた遺伝子構造予測法としてHelixerが開発されました。当初は遺伝子構造を考慮していませんでしたが、後にHMMベースのポストプロセッサを追加することで、最も正確な ab initio予測ツールとされています。しかし、エクソンや遺伝子の予測精度を示す一般的な基準での評価は行われていません。
新たに開発されたTiberiusは、ab initioの哺乳類ゲノムアノテーションでHelixerを上回るパフォーマンスを示しました (遺伝子レベルのF1スコアは55% vs 19%)。
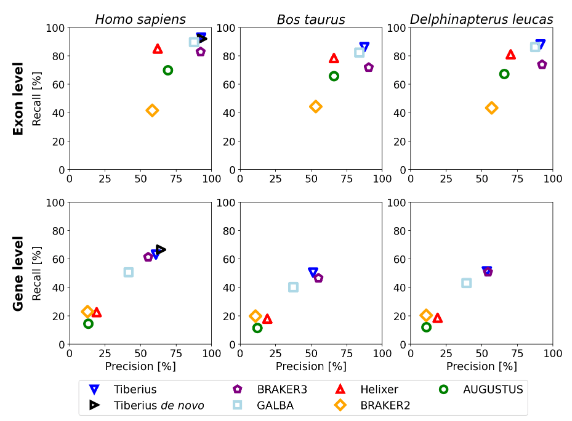
Fig.3:Tiberius、BRAKER3、GALBA、Helixer、BRAKER2、AUGUSTUS の遺伝子およびExonレベルの精度と再現率
さらに、GPUを用いることでHelixerや従来のCPUベースのツールと比較して、大幅な高速化と高い並列処理能力を実現しています。
また、RNAseqとタンパク質配列データを統合する最新ツールのBRAKER3と比較しても、Tiberiusは外部データを使用せずに僅かながら高いパフォーマンスを発揮しました (ただし、Tiberiusは選択的スプライシングを考慮していないことに注意)。
インストール
ソースからのインストールとSingularityコンテナによるインストールが可能です。
Singularityコンテナでインストール (latestだとバージョンがすぐにわからないのでdockerhubの情報よりバージョンをファイル名に記載)
singularity build --fakeroot tiberius_v1.1.1.sif docker://larsgabriel23/tiberius:latest
重み付けデータをダウンロードします。
wget https://bioinf.uni-greifswald.de/bioinf/tiberius/models/tiberius_weights.tgz
tar -xzvf tiberius_weights.tgz
対象種の配列データの取得
哺乳類としてMouseのリファレンスゲノムGRCm39(mm39)を使用します。
EnsemblかNCBIどちらでも良いのでfastaファイルをダウンロードします。今回はNCBIの配列データを使用。
mkdir ref
cd ref & wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/635/GCF_000001635.27_GRCm39/GCF_000001635.27_GRCm39_genomic.fna.gz
解凍します。
pigz -d -p 12 ref/GCF_000001635.27_GRCm39_genomic.fna.gz
scaffoldと記載のない配列情報だけ残し、不要な情報を削除します。
seqkit grep -nrp "scaffold" -v ref/GCF_000001635.27_GRCm39_genomic.fna | \
sed -e "s/,_GRCm39//g" -e "s/\//_/g" > ref/GCF_000001635.27_GRCm39_chr.fna
Tiberiusはリピート領域をsoftmaskしたゲノム配列に対しての使用を推奨しています。softmaskの方法は以下のページを参考にしてください。
RepeatModeler2 → RepeatMasker or Tandem Repeat Finderといった一般的な方法のようで、使用するリファレンスゲノムはソフトマスクされているのでそのまま使います。
Tiberiusの実行
Tiberius USAGE
usage: tiberius.py [-h] [--model_lstm MODEL_LSTM] [--model_hmm MODEL_HMM] [--model MODEL] [--out OUT] --genome GENOME
[--parallel_factor PARALLEL_FACTOR] [--no_softmasking] [--clamsa CLAMSA] [--codingseq CODINGSEQ] [--protseq PROTSEQ] [--emb]
[--strand STRAND] [--seq_len SEQ_LEN] [--batch_size BATCH_SIZE]
Tiberius predicts gene structures from a nucleotide sequences that can have repeat softmasking. There are flexible configuration to load the
model, including options to: - Load a complete LSTM+HMM model - Load only the LSTM model and use a default HMM layer Example usage: Load
LSTM+HMM model: tiberius.py --genome genome.fa --model model_keras_save --out tiberius.gtf Load only LSTM model: tiberius.py --genome genome.fa
--model_lstm lstm_keras_save Load LSTM model and custom HMM Layer: tiberius.py --genome genome.fa --model_lstm lstm_keras_save --model_hmm
hmm_layer_keras_save Use Tiberius with softmasking disabled: tiberius.py --genome genome.fa --model model_keras_save --out tiberius.gtf
--no_softmasking
optional arguments:
-h, --help show this help message and exit
--model_lstm MODEL_LSTM
LSTM model file that can be used with --model_hmm to add a custom HMM layer, otherwise a default HMM layer is added.
--model_hmm MODEL_HMM
HMM layer file that can be used with --model_lstm.
--model MODEL LSTM model file with HMM Layer.
--out OUT Output GTF file with Tiberius gene prediction.
--genome GENOME Genome sequence file in FASTA format.
--parallel_factor PARALLEL_FACTOR
Parallel factor used in Viterbi. Use the factor of w_size that is closest to sqrt(w_size) (817 works well for 500004)
--no_softmasking Disables softmasking.
--clamsa CLAMSA
--codingseq CODINGSEQ
Ouputs the coding sequences of all predicted genes as a FASTA file.
--protseq PROTSEQ Ouputs the amino acid sequences of all predicted genes as a FASTA file.
--emb Indicates if the HMM layer uses embedding input. Currently not supported
--strand STRAND Either "+" or "-" or "+,-".
--seq_len SEQ_LEN Length of sub-sequences used for parallelizing the prediction.
--batch_size BATCH_SIZE
Number of sub-sequences per batch.
自身のPCに搭載されているGPUのメモリに合わせて--batch_sizeオプションを設定します。メモリサイズに対する推奨バッチサイズは以下の通りです。
| GPU | Memory | batch size |
|---|---|---|
| A100 | 80GB | 16 |
| RTX 3090 | 25GB | 8 |
| RTX 2070 | 8GB | 2 |
私はRTX 3070Tiでメモリが8GBなので--batch_size 2を実行オプションに指定します。
推定されたCDS領域のDNA配列データとタンパク質の配列データも同時に出力するには、--codingseqと--protseqを指定します。
最終的な実行コマンド
singularity exec --nv tiberius_v1.1.1.sif \
tiberius.py \
--genome ref/GCF_000001635.27_GRCm39_chr.fna \
--out GRCm39_tiberius_chr.gtf \
--model tiberius_weights \
--batch_size 2 \
--codingseq GRCm39_tiberius_chr_CDS.fna \
--protseq GRCm39_tiberius_chr_CDS.faa \
2>&1 | tee tiberius_20241130.log
最終的には8GBしかない3070tiでは実行できなかったので、少しずるいですがメモリに余裕のあるGPUサーバーを使いました。batch_sizeが16でおよそ1.5時間で全工程が終了しました。
tiberiusの出力
カレントディレクトリに3ファイルが出力されます。
- GRCm39_tiberius_chr.gtf
- GRCm39_tiberius_chr_CDS.fna
- GRCm39_tiberius_chr_CDS.faa
fnaは塩基配列、faaはアミノ酸で推定されたコーディング領域の情報になります。
出力されたgtfはこんな感じです。
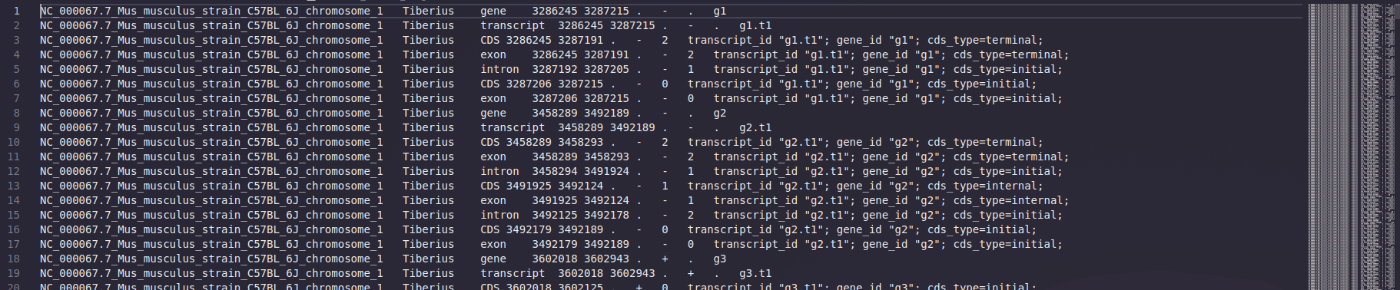
featureにはgene, transcript, CDS, exon, intronの情報が記載されています。
awk -F'\t' '!a[$3]++ {print $3}' GRCm39_tiberius_chr.gtf
gene
transcript
CDS
exon
intron
とりあえ遺伝子定義ファイルとCDS領域の塩基配列、アミノ酸配列情報を得ることができたので終了。
関連記事
Discussion