ロングリードメタゲノムのビニング
こちらの記事で生成したde novoアセンブル後のコンティグ配列のビニングを実施します。
全体の作業イメージは以下のような感じで、今回はビニングの部分です。
Binningとは?
Binningはメタゲノムアセンブルゲノム(MAG)を作成する際に、同じゲノム由来と予測される配列をグルーピングする操作です。

https://onlinelibrary.wiley.com/doi/full/10.1002/mbo3.1298
アンプリコンシーケンスのOTUクラスタリングのようなイメージですね。
各種Binner, das_tool, minimap2のインストール
Binner : SemiBin2, MaxBin2
Binner用の仮想環境 (binning) を作成して、Binningに必要なツールをインストールします。
mamba create -n binning -c conda-forge -c bioconda semibin maxbin2 minimap2 das_tool -y
# Activate virtual env
mamba activate binning
SemiBin2
semibin2 : semibin2は自己教師学習によるBinnerです。
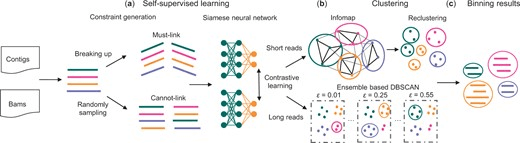
https://academic.oup.com/bioinformatics/article/39/Supplement_1/i21/7210480
SemiBin2 USAGE
usage: SemiBin2 [-h] [-v] [--verbose | --quiet] ...
Neural network-based binning of metagenomic contigs
optional arguments:
-h, --help show this help message and exit
-v, -V, --version Print the version number
--verbose Verbose output (default: False)
--quiet, -q Quiet output (default: False)
SemiBin subcommands:
single_easy_bin Bin contigs (single or co-assembly) using one command.
multi_easy_bin Bin contigs (multi-sample mode) using one command.
generate_cannot_links (predict_taxonomy)
Run the contig annotation using mmseqs with GTDB reference genome and generate cannot-link file used in the
semi-supervised deep learning model training. This will download the GTDB database if not downloaded before.
generate_sequence_features_single
Generate sequence features (kmer and abundance) as training data for (semi/self)-supervised deep learning model
training (single or co-assembly mode). This will produce the data.csv and data_split.csv files.
generate_sequence_features_multi (generate_sequence_features_multi)
Generate sequence features (kmer and abundance) as training data for (semi/self)-supervised deep learning model
training (multi-sample mode). This will produce the data.csv and data_split.csv files.
download_GTDB Download GTDB reference genomes.
check_install Check whether required dependencies are present.
concatenate_fasta concatenate fasta files for multi-sample binning
train_semi Train the model.
train_self Train the model with self-supervised learning
bin Group the contigs into bins.
bin_long Group the contigs from long reads into bins.
For more information, see https://semibin.readthedocs.io/en/latest/subcommands/
MaxBin2
複数のメタゲノムシーケンシングリードを同時アセンブリした後、contigのテトラヌクレオチド頻度と、関連するすべてのメタゲノムリードのカバレッジを測定し、contigを個々のbinに分類。Binの存在量をExpectation-Maximization (EM) アルゴリズムで推定します。

https://academic.oup.com/bioinformatics/article/32/4/605/1744462
MaxBin2 USAGE
MaxBin 2.2.7
Unrecognized token [--help]
MaxBin - a metagenomics binning software.
Usage:
run_MaxBin.pl
-contig (contig file)
-out (output file)
(Input reads and abundance information)
[-reads (reads file) -reads2 (readsfile) -reads3 (readsfile) -reads4 ... ]
[-abund (abundance file) -abund2 (abundfile) -abund3 (abundfile) -abund4 ... ]
(You can also input lists consisting of reads and abundance files)
[-reads_list (list of reads files)]
[-abund_list (list of abundance files)]
(Other parameters)
[-min_contig_length (minimum contig length. Default 1000)]
[-max_iteration (maximum Expectation-Maximization algorithm iteration number. Default 50)]
[-thread (thread num; default 1)]
[-prob_threshold (probability threshold for EM final classification. Default 0.9)]
[-plotmarker]
[-markerset (marker gene sets, 107 (default) or 40. See README for more information.)]
(for debug purpose)
[-version] [-v] (print version number)
[-verbose]
[-preserve_intermediate]
Please specify either -reads or -abund information.
You can input multiple reads and/or abundance files at the same time.
Please read README file for more details.
DAS_tool
DAS_Tool (Dereplication, Aggregation, Scoring Tool)は、複数のBinningアルゴリズムの結果を統合して、単一のアセンブリから最適化されたBinのセットを自動的に計算するツールです。
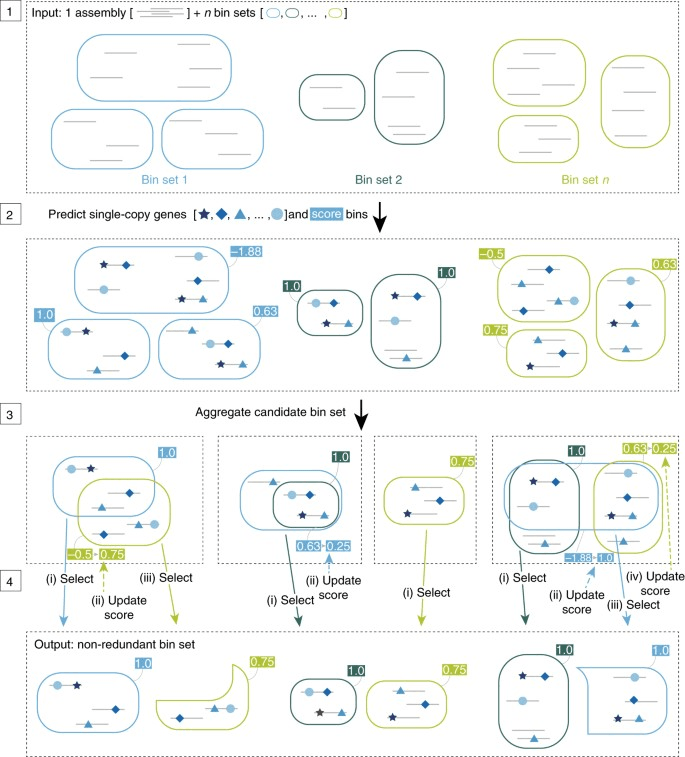
https://www.nature.com/articles/s41564-018-0171-1
DAS_Tool USAGE
DAS Tool version 1.1.2
Usage: DAS_Tool -i methodA.scaffolds2bin,...,methodN.scaffolds2bin
-l methodA,...,methodN -c contigs.fa -o myOutput
-i, --bins Comma separated list of tab separated scaffolds to bin tables.
-c, --contigs Contigs in fasta format.
-o, --outputbasename Basename of output files.
-l, --labels Comma separated list of binning prediction names. (optional)
--search_engine Engine used for single copy gene identification [blast/diamond/usearch].
(default: usearch)
--write_bin_evals Write evaluation for each input bin set [0/1]. (default: 1)
--create_plots Create binning performance plots [0/1]. (default: 1)
--write_bins Export bins as fasta files [0/1]. (default: 0)
--write_unbinned Report unbinned contigs. To export as fasta file also set write_bins==1 [0/1]. (default: 0)
--proteins Predicted proteins in prodigal fasta format (>scaffoldID_geneNo).
Gene prediction step will be skipped if given. (optional)
-t, --threads Number of threads to use. (default: 1)
--score_threshold Score threshold until selection algorithm will keep selecting bins [0..1].
(default: 0.5)
--duplicate_penalty Penalty for duplicate single copy genes per bin (weight b).
Only change if you know what you're doing. [0..3]
(default: 0.6)
--megabin_penalty Penalty for megabins (weight c). Only change if you know what you're doing. [0..3]
(default: 0.5)
--db_directory Directory of single copy gene database. (default: install_dir/db)
--resume Use existing predicted single copy gene files from a previous run [0/1]. (default: 0)
--debug Write debug information to log file.
-v, --version Print version number and exit.
-h, --help Show this message.
Example 1: Run DAS Tool on binning predictions of MetaBAT, MaxBin, CONCOCT and tetraESOMs. Output files will start with the prefix DASToolRun1:
DAS_Tool -i sample_data/sample.human.gut_concoct_scaffolds2bin.tsv,sample_data/sample.human.gut_maxbin2_scaffolds2bin.tsv,sample_data/sample.human.gut_metabat_scaffolds2bin.tsv,sample_data/sample.human.gut_tetraESOM_scaffolds2bin.tsv -l concoct,maxbin,metabat,tetraESOM -c sample_data/sample.human.gut_contigs.fa -o sample_output/DASToolRun1
Example 2: Run DAS Tool again with different parameters. Use the proteins predicted in Example 1 to skip the gene prediction step. Set the number of threads to 2 and score threshold to 0.1. Output files will start with the prefix DASToolRun2:
DAS_Tool -i sample_data/sample.human.gut_concoct_scaffolds2bin.tsv,sample_data/sample.human.gut_maxbin2_scaffolds2bin.tsv,sample_data/sample.human.gut_metabat_scaffolds2bin.tsv,sample_data/sample.human.gut_tetraESOM_scaffolds2bin.tsv -l concoct,maxbin,metabat,tetraESOM -c sample_data/sample.human.gut_contigs.fa -o sample_output/DASToolRun2 --threads 2 --score_threshold 0.6 --proteins sample_output/DASToolRun1_proteins.faa
Please cite: Sieber et al., 2018, Nature Microbiology (https://doi.org/10.1038/s41564-018-0171-1).
これで実行準備ができました。
Binning
hifiasm_meta contig
SemiBin2にHiFi readをcontigにmappingしたBAMファイルが必要なのでminimap2を使ってmappingします。
Note : SemiBinのtutorialではbowtie2でBamを作成していた。
# Run
minimap2 \
-t 30 \
-ax map-hifi \
02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta.gz \
01fq/m64011_210224_000525.hifi_reads.fastq.gz | \
samtools sort -@ 30 -O BAM - > 02asm_meta/m64011_210224_000525.hifi_reads.bam && \
samtools index -@ 30 02asm_meta/m64011_210224_000525.hifi_reads.bam
SemiBin2によるBinning
# output directoryの作成
mkdir 02asm_meta/semibin2
# Run
SemiBin2 single_easy_bin \
--sequencing-type=long_read \
-i 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta.gz \
-b 02asm_meta/m64011_210224_000525.hifi_reads.bam \
-o 02asm_meta/semibin2/ \
--environment human_gut \
--engine cpu \
2>&1 | tee runlog/hifiasm_metaSemiBin2.log
MaxBin2によるBinning
# 出力フォルダ
mkdir 02asm_meta/maxbin2
pigz -d 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta.gz
# Run
run_MaxBin.pl \
-contig 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta \
-reads 01fq/m64011_210224_000525.hifi_reads.fastq.gz \
-out 02asm_meta/maxbin2/maxbin2 \
-thread 20 \
2>&1 | tee runlog/hifiasm_metaMaxBin2.log
出力ファイルと解凍したコンティグ配列ファイルを圧縮しておきます。
pigz 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta
pigz 02asm_meta/maxbin2/*
metaFlye
hifiasm-metaと同様にmetaFlyeもminimap2でHiFi readをマッピングします。
# Run
minimap2 \
-t 30 \
-ax map-hifi \
02metaFlye/assembly.fasta.gz \
01fq/m64011_210224_000525.hifi_reads.fastq.gz | \
samtools sort -@ 30 -O BAM - > 02metaFlye/m64011_210224_000525.hifi_reads.bam && \
samtools index -@ 30 02metaFlye/m64011_210224_000525.hifi_reads.bam
10分くらいで終わりました。
SemiBin2によるBinning
# output directoryの作成
mkdir 02metaFlye/semibin2
# Run
SemiBin2 single_easy_bin \
--sequencing-type=long_read \
-i 02metaFlye/assembly.fasta.gz \
-b 02metaFlye/m64011_210224_000525.hifi_reads.bam \
-o 02metaFlye/semibin2/ \
--environment human_gut \
--engine cpu \
2>&1 | tee runlog/metaFlyeSemiBin2.log
MaxBin2によるBinning
# 出力フォルダ
mkdir 02metaFlye/maxbin2
pigz -d 02metaFlye/assembly.fasta.gz
# Run
run_MaxBin.pl \
-contig 02metaFlye/assembly.fasta \
-reads 01fq/m64011_210224_000525.hifi_reads.fastq.gz \
-out 02metaFlye/maxbin2/maxbin2 \
-thread 30 \
2>&1 | tee runlog/metaFlyeMaxBin2.log
出力ファイルと解凍したコンティグ配列ファイルを圧縮しておきます。
pigz 02metaFlye/assembly.fasta
pigz 02metaFlye/maxbin2/*
metaMDBG
最後はmetaMDBGです。minimap2でHiFi readをマッピングします。
# Run
minimap2 \
-t 30 \
-ax map-hifi \
02metaDBG/m64011_210224_000525.hifi_reads.contigs.fasta.gz \
01fq/m64011_210224_000525.hifi_reads.fastq.gz | \
samtools sort -@ 30 -O BAM - > 02metaDBG/m64011_210224_000525.hifi_reads.bam && \
samtools index -@ 30 02metaDBG/m64011_210224_000525.hifi_reads.bam
SemiBin2によるBinning
# output directoryの作成
mkdir 02metaMDBG/semibin2
# Run
SemiBin2 single_easy_bin \
--sequencing-type=long_read \
-i 02metaMDBG/contigs.fasta.gz \
-b 02metaMDBG/m64011_210224_000525.hifi_reads.bam \
-o 02metaMDBG/semibin2/ \
--environment human_gut \
--engine cpu \
2>&1 | tee runlog/metaMDBGSemiBin2.log
MaxBin2によるBinning
# 出力フォルダ
mkdir 02metaMDBG/maxbin2
pigz -d 02metaMDBG/contigs.fasta.gz
# Run
run_MaxBin.pl \
-contig 02metaMDBG/contigs.fasta \
-reads 01fq/m64011_210224_000525.hifi_reads.fastq.gz \
-out 02metaMDBG/maxbin2/maxbin2 \
-thread 30 \
2>&1 | tee runlog/metaMDBGMaxBin2.log
出力ファイルと解凍したコンティグ配列ファイルを圧縮しておきます。
pigz 02metaMDBG/contigs.fasta
pigz 02metaMDBG/maxbin2/*
ここまでのツールの実行時間とInputしたcontig数以下のとおりです。
| SemiBin2 | Maxbin2 | Inputしたコンティグ数 | |
|---|---|---|---|
| hifiasm-meta | 21 | 46 | 11,693 |
| metaFlye | 22 | 41 | 10,419 |
| metaMDBG | 22 | 52 | 26,769 |
DAS_tool
DAS_toolにinputするtsvファイルをFasta_to_Contig2Bin.shで使って生成します。
DAS_toolのリポジトリをクローンして、各アセンブラーごとにFasta_to_Contig2Bin.shを実行します。
git clone https://github.com/cmks/DAS_Tool.git
hifiasm-meta
容量削減のために圧縮していたファイルを解凍します。
#解凍
pigz -d 02asmmeta/semibin2/output_bins/*.fa.gz
pigz -d 02asm_meta/maxbin2/*.fasta.gz
Fasta_to_Contig2Binにbinning後の配列データが含まれているファイルと拡張子を渡すこと、各コンティグ配列名称とBin名称との対応表が生成されます。
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02asm_meta/semibin2/output_bins/ -e fa > 02asm_meta/semibin2.tsv
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02asm_meta/maxbin2 -e fasta > 02asm_meta/maxbin2.tsv
こんな感じ。
| Contig配列名称 | Bin名称 |
|---|---|
| s344.ctg000546c | SemiBin_0 |
| s116.ctg000175l | SemiBin_100 |
| s1423.ctg002027l | SemiBin_101 |
| ... | ... |
| Contig配列名称 | Bin名称 |
|---|---|
| s76.ctg000116c | maxbin2.001 |
| s1512.ctg002138l | maxbin2.001 |
| s8346.ctg009811l | maxbin2.001 |
| ... | ... |
終わったらまた圧縮しときます。
pigz 02asm_meta/semibin2/output_bins/*.fa
pigz 02asm_meta/maxbin2/*.fasta
DAS_Tool \
--bins 02asm_meta/semibin2.tsv,02asm_meta/maxbin2.tsv \
--contigs 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta \
--labels semibin2,maxbin2 \
--threads 30 \
--outputbasename 02asm_meta/DASTool/DASTool \
--write_bins 1 \
--search_engine diamond 2>&1 | tee runlog/hifiasm-meta_dastool.log
metaFlye
容量削減のために圧縮していたファイルを解凍します。
pigz -d 02metaFlye/semibin2/output_bins/*.fa.gz
pigz -d 02metaFlye/maxbin2/*.fasta.gz
対応表の作成
# Fasta_to_Contig2Bin.sh
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02metaFlye/semibin2/output_bins/ -e fa > 02metaFlye/semibin2.tsv
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02metaFlye/maxbin2 -e fasta > 02metaFlye/maxbin2.tsv
終わったら圧縮。
pigz 02metaFlye/semibin2/output_bins/*.fa
pigz 02metaFlye/semibin2/output_bins/*.fa
DAS_Tool \
--bins 02metaFlye/maxbin2.tsv,02metaFlye/semibin2.tsv \
--contigs 02metaFlye/assembly.fasta \
--labels semibin2,maxbin2 \
--threads 30 \
--outputbasename 02metaFlye/DASTool/DASTool \
--write_bins 1 \
--search_engine diamond 2>&1 | tee runlog/metaFlye_dastool.log
metaMDBG
pigz -d 02metaMDBG/semibin2/output_bins/*.fa
pigz -d 02metaMDBG/maxbin2/*.fasta
# contig配列を解凍
pigz -d 02metaMDBG/contigs.fasta.gz
対応表の作成
# Fasta_to_Contig2Bin.sh
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02metaMDBG/semibin2/output_bins/ -e fa > 02metaMDBG/semibin2.tsv
bash DAS_Tool/src/Fasta_to_Contig2Bin.sh -i 02metaMDBG/maxbin2 -e fasta > 02metaMDBG/maxbin2.tsv
DAS_Tool \
--bins 02metaMDBG/maxbin2.tsv,02metaMDBG/semibin2.tsv \
--contigs 02metaMDBG/contigs.fasta \
--labels semibin2,maxbin2 \
--threads 30 \
--outputbasename 02metaMDBG/DASTool/DASTool \
--write_bins 1 \
--search_engine diamond 2>&1 | tee runlog/metaMDBG_dastool.log
終わったら圧縮
pigz 02metaMDBG/semibin2/output_bins/*.fa
pigz 02metaMDBG/maxbin2/*.fasta
pigz 02metaMDBG/contigs.fasta.gz
これらの処理でコンティグ配列を分類することができました。
思ったより長くなってきたので今回はここまで。
次はCheckM2でBinの品質確認です。
Information
- 自作PC1
- minimap2 :
- DAS Tool : 1.1.2
- MaxBin : 2.2.7
- SemiBin2 :
Discussion