ロングリードメタゲノムアセンブラー三兄弟の比較 : hifiasm_meta, metaMDBG, metaFlye
ショットガンメタゲノムのメジャーなロングリードアセンブラーを比較します。
ロングリードのメタゲノムアセンブリ

https://www.sciencedirect.com/science/article/pii/S2001037021004931#f0010
ショットガンメタゲノム解析では、配列決定したリードを元の微生物ゲノムに組み上げていく作業を指します(上記図B左列上段)。
シーケンサーで読むために物理的に剪断したDNA配列をアセンブルすることで、微生物ゲノムの遺伝子情報やコミュニティ構造、多様性に関する情報を得ることができます。
メタゲノムロングリードシーケンス
近年、Pacific Biosciences (PacBio) や Oxford Nanopore Technologies (ONT)といった第3世代シーケンサーが得意とするロングリードシーケンスの分析コストが低くなってきています。
配列決定精度の向上や、2022,2023年の新機種発売によるスループット向上が主な理由だと思います。
また、ショートリードをメインにしていたIlluminaもComplete Long Readを発表してきたことから、徐々にロングリード時代への移り変わりが活発になってきていると言っても過言ではないでしょう。
ロングリードシーケンスは、ショートリードシーケンスと比較して、微生物ゲノムの再構築の難易度を大きく下げてくれます。
一方、ロングリードとはいえ環境サンプルでは、種の不均一性やゲノム内の長鎖リピート配列などの影響により、単離株やヒトのゲノムアセンブルと比較すると計算上の課題が発生します。
シーケンサーによって得手不得手はありますが、私が扱う頻度が高いPacBioのHiFi readを使ってmetaFlye, HiFiasm-meta, metaMDBGの3つのアセンブラーを動作させて見たいと思います。
ロングリードメタゲノムアセンブリツール
metaFlye
-
preprint : https://www.biorxiv.org/content/10.1101/637637v1.full
-
Flye and metaFlye: algorithms for long-read de novo assembly using repeat graphs
(solid k-mersとリピートグラフを利用したアセンブルと理解していますが、Flye/metaFlyeは使わないのでよくわかりません...)
hifiasm-meta
2022年に発表されたアセンブラーです。ロングリードメタゲノムアセンブラーがNanopore用のmetaFlyeしか公開されていなかったため、hifiasmをメタゲノムサンプル用に拡張したhifiasm-metaを開発したとの記載がありました。
また、2023年に公開された上記プレプリントでは、PacBioのHiFiリードに対して、11のDe novoアッセンブリーツール(HiCanu, hifiasm, HiFlye, hifiasm-meta, metaFlye, Peregrine, Shasta, Verkko, MECAT2, miniasm, NextDenovo)の包括的なベンチマークを行っています。
-
真核生物 : HiCanu, hifiasm, HiFlye, Peregrine, Shasta, Verkko, MECAT2, miniasm, NextDenovo
-
原核生物 : HiCanu, hifiasm-meta, metaFlye, NextDenovo
総評として、真核生物であればhifiasm、腸内細菌ではhifiasm-metaが総合的に良い結果を出していました。
metaMDBG
2024/1/2更新 ※プレプリントはこちら
minimizer-space assemblyの原理を採用した手法(*1,*2)です。Assembly結果のフィードバックを行うことでk'-min-mersのサイズを増加させるmulti-k'アプローチを設計し、メタゲノムデータのアセンブリに適用します(難しい)。
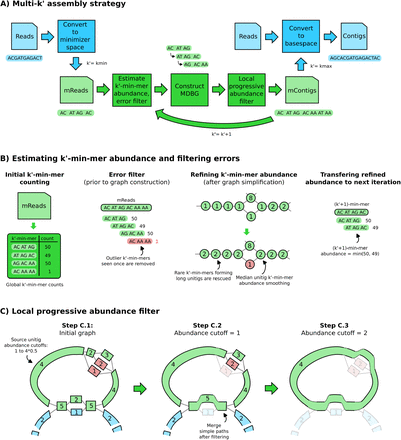
他のアセンブラと比較して、1.5 ~ 12倍高速で、メモリ (RAM) の使用量が 1/10 ~ 1/130 と高速でかつ低メモリなアセンブラーです。
*1 : 類似したアセンブラーとしてrust-mdbgがありますが、メタゲノム向けに設計されていません
*2 : 低いシーケンスエラーのロングリードデータに対して適用される手法
比較用のデータセット
データはPacBioのftpサイトよりダウンロードしたものを使います。Sequel lleでシーケンスされたデータです。
- m64011_210225_094432 - The BioCollective pooled gut microbiome sample 138390; 9.2kb insert
mkdir 01fq
aria2c -x 5 -ctrue https://downloads.pacbcloud.com/public/dataset/Sequel-IIe-202104/metagenomics/m64011_210225_094432.hifi_reads.fastq.tar.gz -d 01fq
作業ステップは以下のようなイメージです。
では、行きましょう。
仮想環境&実行環境の設定
pb-metaという仮想環境を作成します。python3のバージョンは3.8.16にしました。
mamba create -n pb-meta python=3.8.16 -y
mamba activate pb-meta
以降は、特筆しなければこの仮想環境内を使って作業を進めます。
また、実行ログを集めるフォルダも作っておきます。
mkdir runlog
De novo assebly
hifiasm-meta
mambaを使ってインストールします。
mamba install -c bioconda hifiasm_meta -y
hifiasm-meta USAGE
Usage: hifiasm_meta 0.3-r063.2 (hifiasm code base 0.13-r308)
Options:
Input/Output:
-o STR prefix of output files [hifiasm_meta.asm]
-B STR prefix of bin files, if it's different from -o [hifiasm_meta.asm]
-i ignore saved read correction and overlaps
-t INT number of threads [1]
-z INT length of adapters that should be removed [0]
--version show version number
Read selection:
--force-preovec
enable and force read selection.
--lowq-10
lower 10% runtime kmer frequency threshold. [50]
--lowq-5
lower 5% runtime kmer frequency threshold. [50]
--lowq-3
lower 3% runtime kmer frequency threshold. [10]
Overlap/Error correction:
-k INT k-mer length (must be <64) [51]
-w INT minimizer window size [51]
-f INT number of bits for bloom filter; 0 to disable [37]
-D FLOAT drop k-mers occurring >FLOAT*coverage times [5.0]
-N INT consider up to max(-D*coverage,-N) overlaps for each oriented read [100]
-r INT round of correction [3]
Assembly:
-a INT round of assembly cleaning [4]
-n INT remove tip unitigs composed of <=INT reads [3]
-x FLOAT max overlap drop ratio [0.8]
-y FLOAT min overlap drop ratio [0.2]
Auxiliary:
-e ban assembly, i.e. terminate before generating string graph
--write-paf dump overlaps (paf).
--dump-all-ovlp
dump all overlaps ever calculated in the final overlapping (paf).
--write-ec
dump error corrected reads (fasta).
Example: ./hifiasm_meta -o asm -t 32 asm.fq.gz 2>log
See `man ./hifiasm_meta.1` for detailed descriptions command-line options.
mkdir 02asm_meta
time hifiasm_meta -t30 -o 02asm_meta/m64011_210224_000525.hifi_reads ./01fq/m64011_210224_000525.hifi_reads.fastq.gz 2> ./runlog/hifiasm-meta.log
標準出力はrunlogフォルダにhifiasm-meta.logとして出力されるようにしてあります。
1サンプルにつき16ファイル出力されますが、プライマリコンティグの配列が含まれているm64011_210224_000525.hifi_reads.p_ctg.gfaを使います。
配列データを抽出します。
awk '/^S/{print ">"$2;print $3}' 02asm_meta/m64011_210224_000525.hifi_reads.p_ctg.gfa > 02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta
大きいので圧縮しておきます。
find ./02asm_meta -name '*.fa' -type f -execdir pigz {} \;
find ./02asm_meta -name '*.fasta' -type f -execdir pigz {} \;
find ./02asm_meta -name '*.gfa' -type f -execdir pigz {} \;
metaFlye
mambaを使ってインストールします。
mamba install -c bioconda flye -y
metaFlye USAGE
usage: flye (--pacbio-raw | --pacbio-corr | --pacbio-hifi | --nano-raw |
--nano-corr | --nano-hq ) file1 [file_2 ...]
--out-dir PATH
[--genome-size SIZE] [--threads int] [--iterations int]
[--meta] [--polish-target] [--min-overlap SIZE]
[--keep-haplotypes] [--debug] [--version] [--help]
[--scaffold] [--resume] [--resume-from] [--stop-after]
[--read-error float] [--extra-params]
[--deterministic]
flye: error: the following arguments are required: -o/--out-dir
ゲノムサイズはデフォルト
mkdir 02metaFlye
time flye \
--pacbio-hifi 01fq/m64011_210224_000525.hifi_reads.fastq.gz \
-o 02metaFlye \
--genome-size "392m" \
--threads=30 \
2>&1 | tee ./runlog/metaflye.log
こちらも、標準出力はrunlogフォルダにmetaflye.logとして出力されるようにしてあります。
metaMDBG
GithubのInstallationの指示に従ってインストールしました。
# Download metaMDBG repository
git clone https://github.com/GaetanBenoitDev/metaMDBG.git
# Create metaMDBG conda environment
cd metaMDBG
mamba env create -f conda_env.yml -y
mamba activate metaMDBG
mamba env config vars set CPATH=${CONDA_PREFIX}/include:${CPATH}
mamba deactivate
# Activate metaMDBG environment
mamba activate metaMDBG
# Compile the software
mkdir build
cd build
cmake ..
make -j 30
インストールが成功すると、metaMDBG という名前の実行可能ファイルが /build/binフォルダに出力されます。
毎回相対pathを記述するのは面倒なので、一時的にpathを登録しておきます。
export PATH=$PATH:${PWD}/build/bin
metaMDBG USAGE
Program: metaMDBG (assembly of long and accurate metagenomics reads)
Version: 0.3
Contact: Gaëtan Benoit (gaetanbenoitdev@gmail.com)
Usage: metaMDBG [command]
command:
asm : perform read assembly
polish : polish contigs
derep : purge strain duplication
gfa : generate an assembly graph (.gfa). Require a finished metaMDBG run
time metaMDBG asm 02metaMDBG/ 01fq/m64011_210224_000525.hifi_reads.fastq.gz -t 30 2>&1 | tee ./runlog/metaMDBG.log
OUTPUTフォルダ、HiFi readの順に指定します。
Results
summary
それぞれのアセンブラの実行に関する結果です。
| run time (min) | CPU time (min) | Peak RAM (GB) | |
|---|---|---|---|
| hifiasm_meta | 323 | 9025 | 74.7 |
| metaflye | 162 | 3793 | - |
| metaMDBG | 147 | 2279 | 6.36 |
各アセンブラが出力したコンティグについてもseqkitで確認しました。
| Num seqs | min len | max len | ave len | N50 | |
|---|---|---|---|---|---|
| hifiasm_meta | 11,693 | 4,918 | 4,413,267 | 98,364.8 | 302,611 |
| metaflye | 10,419 | 518 | 4,412,735 | 96,945.0 | 181,422 |
| metaMDBG | 26,769 | 510 | 7,383,977 | 43,713.0 | 201,253 |
metaMDBGが前評判通り実行時間は一番短く、メモリパフォーマンスも高かったです。ただ一方、N50はhifiasm_metaが最も長く、それに次いで100kbp程度いmetaMDBG, metaFlyeという結果でした。
contig length frequency
contig配列の分布が気になったので、頻度分布のdensity plotを以下のRスクリプトで書いてみました。
plot Rscript
# load package
require(seqinr)
require(ggplot2)
require(dplyr)
# import contig
hifiasm_meta_contig <- seqinr::read.fasta("02asm_meta/m64011_210224_000525.hifi_reads.contigs.fasta")
metaFlye_contig <- seqinr::read.fasta("02metaFlye/assembly.fasta")
metaMDBG_contig <- seqinr::read.fasta("02metaMDBG/contigs.fasta")
# Obtain contig length summary
hifiasm_sequence_lengths <- getLength(hifiasm_meta_contig)
metaFlye_sequence_lengths <- getLength(metaFlye_contig)
metaMDBG_sequence_lengths <- getLength(metaMDBG_contig)
# Convert to DataFrame
hifiasm_length_data <- data.frame(Length = hifiasm_sequence_lengths) %>%
dplyr::mutate(tool = "hifiasm")
metaFlye_length_data <- data.frame(Length = metaFlye_sequence_lengths) %>%
dplyr::mutate(tool = "metaFlye")
metaMDBG_length_data <- data.frame(Length = metaMDBG_sequence_lengths) %>%
dplyr::mutate(tool = "metaMDBG")
# concat DataFrame
length_data <- dplyr::bind_rows(hifiasm_length_data, metaFlye_length_data, metaMDBG_length_data)
# Density plot
ggplot(length_data, aes(x = Length, fill = tool, color = tool)) +
geom_density(alpha=0.5) +
xlim(c(0, 2.0e+5)) +
labs(title = "Distribution of contig length", x = "Contig Length", y = "Freqency")

最大配列帳は2Mbpで切ってあります。hifiasm-metaとmetaFlyeはmetaMDBGより長めでほぼ同等のcontig配列長にピークがあります。metaFlyeは裾の尾が長めな分布していますね。
metaMDBGはhifiasm-meta, metaFlyeと比較すると短い位置にピークがあります。
これらの違いが最終的なプロファイルにどう関わってくるか気になるところです。
次は、SemiBin2, MaxBin2を使ってBinningしてMAGを生成後、DAS_toolsでBinning結果を統合する予定です。
↓ 記事を公開しました。
Information
- 自作PC1
- metaMDBG : v0.3
- metaFlye : 2.9.2-b1786
- hifiasm-meta : ha base version: 0.13-r308, hamt version: 0.3-r063.2
Discussion