多機能なメタゲノム解析パイプライン : SqueezeMeta (+α SQMtools)
はじめに
ショットガンメタゲノム解析はマイクロバイオームの構造と機能を分析するための標準的な手法となっています。
メタゲノム解析で生成される膨大な数のショートリードを扱うために、多くの新しいバイオインフォマティクスツールとアプローチが開発されています。ただ、それぞれのツールの結果は直接互換性がないことが多く、実質メタゲノム解析は多様なソフトウェアを含む、非標準的な処理で構成される複雑なタスクとなっています。
そんな複雑なタスクをつなぎ合わせて処理するいくつかの標準的なメタゲノムパイプラインには、リードのキュレーション、アセンブリ、遺伝子予測、および結果として得られる遺伝子の機能的および分類学的アノテーションが含まれます。
アセンブリは多くの遺伝子を含む長鎖のゲノム断片を回収することができるため、推奨される解析ステップです。
アセンブルの欠点は、異なるゲノムの一部を誤ってアセンブルすることによるキメラcontigの生成と、存在量が少ない生物種のreadをアセンブル出来ないことがある点です。
低存在量の生物種のreadについては、最初にアセンブルされなかったreadの一部を再Assembleすることでパフォーマンスの向上することが論文として発表されています。
また、本パイプラインで実行することのできるCo-assemblyを実行することでもこの問題を大幅に軽減することもできるようです。
加えて、Co-assemblyはビニングのための最良の戦略です。異なるサンプルのcontigの存在量と組成を比較することで、どのcontigが同じ生物に属するかを決定することができます。
こうして得られた完成度の異なる数十から数百のゲノムビンは、マイクロバイオームの機能をより深く分析するための出発点となります。
SqueezeMeta Intro参照
SqueezeMeta
SqueezeMetaは基本的な解析ステップをカバーするメタゲノミクス/メタトランスクリプトミクス用の完全自動パイプラインです。
ショートリードとロングリードのfastqファイルをinputとしてde novo assemblyやbinning、機能解析が実行できます。

Table1. SqueezeMetaと比較したときの既存メタゲノムパイプラインの特徴
Install via conda
まずは作業場所を作成
mkdir squeezemeta && cd sequeezemeta
作業場所に入ったらsqueezemetaの1.6のインストールと仮想環境を作成
mamba create -n SqueezeMeta -c conda-forge -c bioconda -c anaconda -c fpusan squeezemeta=1.6 --no-channel-priority
USAGE SqueezeMeta
Usage: SqueezeMeta.pl -m <mode> -p <project name> -s <samples file> -f <sequence dir> [options]
Arguments:
Mandatory parameters:
-m <mode>: Mode (sequential, coassembly, merged, seqmerge) (REQUIRED)
-s|-samples <samples file>: Samples file (REQUIRED)
-f|-seq <sequence dir>: fastq/fasta read files' directory (REQUIRED)
-p <project name>: Project name (REQUIRED in coassembly and merged modes)
Restarting
--restart: Restarts the given project where it stopped (project must be speciefied with -p option) (will NOT overwite previous results, unless --force-overwrite is also provided)
-step <step number>: In combination with --restart, restarts the project starting in the given step number (combine with --force_overwrite to regenerate results)
--force_overwrite: Do not check for previous results, and overwrite existing ones
Filtering:
--cleaning: Filters with Trimmomatic (Default: No)
-cleaning_options [options]: Options for Trimmomatic (Default:LEADING:8 TRAILING:8 SLIDINGWINDOW:10:15 MINLEN:30)
Assembly:
-a: assembler <megahit, spades, rnaspades, spades-base, canu, flye> (Default: megahit)
-assembly_options [options]: Extra options to be passed when calling the mapper
-c|-contiglen <size>: Minimum length of contigs (Default: 200)
-extassembly <file>: External assembly, file containing a fasta file of contigs (overrides all assembly steps).
--sg|--singletons: Add unassembled reads to the contig file, as if they were contigs
-contigid <string>: Nomenclature for contigs (Default: assembler´s name)
--norename: Don't rename contigs (Use at your own risk, characters like '_' in contig names will make it crash)
Mapping:
-map: mapping software <bowtie, bwa, minimap2-ont, minimap2-pb, minimap2-sr> (Default: bowtie)
-mapping_options [options]: Extra options to be passed when calling the mapper
ONT support:
--minion: Run on MinION reads (assembler: canu; mapper: minimap2-ont; consensus: 20) (Default: no)
Annotation:
-db <file>: Specify a new taxonomic database
--nodiamond: Check if Diamond results are already in place, and just in that case skips the Diamond run (Default: no)
--nocog: Skip COG assignment (Default: no)
--nokegg: Skip KEGG assignment (Default: no)
--nopfam: Skip Pfam assignment (Default: no)
--euk: Drop identity filters for eukaryotic annotation (Default: no)
-consensus <value>: Minimum percentage of genes for a taxon needed for contig consensus (Default: 50)
--D|--doublepass: First pass looking for genes using gene prediction, second pass using BlastX (Default: no)
-extdb <database file>: List of user-provided databases
-b|-block-size <block size>: block size for diamond against the nr database (Default: calculate automatically)
Binning:
--nobins: Skip all binning (Default: no). Overrides -binners
--onlybins: Run only assembly, binning and bin statistics (including GTDB-Tk if requested) (Default: no)
-binners: Comma-separated list with the binning programs to be used (available: maxbin, metabat2, concoct) (Default: concoct,metabat2)
-taxbinmode <s,c,s+c,c+s>: Source of taxonomy annotation of bins (s: SqueezeMeta; c: CheckM; s+c: SqueezeMeta+CheckM; c+s: CheckM+SqueezeMeta; (Default: s)
--gtdbtk: Run GTDB-Tk to classify the bins. Requires a working GTDB-Tk installation in available in your environment
-gtdbtk_data_path: Path to the GTDB database, by default it is assumed to be present in /path/to/SqueezeMeta/db/gtdb
-extbins: Path to a directory containing external genomes/bins provided by the user. There must be one file per genome/bin, containing each contigs in the fasta format. This overrides the assembly and binning steps
Performance:
-t <threads>: Number of threads (Default: 12)
-canumem <mem>: memory for canu in Gb (Default: 32)
--lowmem: run on less than 16Gb of memory (Default:no)
Other:
-test <step>: Running in test mode, stops AFTER the given step number
--empty: Creates a empty directory structure and conf files, does not run the pipeline
Information:
-v: Version number
-h: This help
インストールが完了する前に、Kronaを使う場合は、ktUpdateTaxonomy.shで分類データベースを手動更新してね。別の場所に保存したい場合はシンボリックリンクを使って変更するように。という表示が出ました。
もし、Krona plotを出力したい場合には、指示に従ってデータベースを準備しましょう。
Krona installed. You still need to manually update the taxonomy
databases before Krona can generate taxonomic reports. The update
script is ktUpdateTaxonomy.sh. The default location for storing
taxonomic databases is <your home dir>/miniconda3/envs/SqueezeMeta/opt/krona/taxonomy
If you would like the taxonomic data stored elsewhere, simply replace
this directory with a symlink. For example:
rm -rf <your home dir>/miniconda3/envs/SqueezeMeta/opt/krona/taxonomy
mkdir /path/on/big/disk/taxonomy
ln -s /path/on/big/disk/taxonomy <your home dir>/miniconda3/envs/SqueezeMeta/opt/krona/taxonomy
ktUpdateTaxonomy.sh
仮想環境が作成できたらActivateする。
mamba activate SqueezeMeta
依存環境の確認
test_install.plを実行すると必要なツールが準備できているか確認してくれます。
test_install.pl
ザーッとpythonやperl, Rの環境などの確認が入ります。
多分この段階ではSqueezeMeta doesn't know where the database are located!とwarningが出ると思うので、案内に従ってDatabaseの準備をします。
Databaseのダウンロード
以下のような出力が得られていたらデータベースの場所をSqueezeMetaが認識できていません。
Checking that SqueezeMeta is properly configured...
SqueezeMeta doesn't know where the databases are located!
If you didn't download them yet please run:
* perl <your home dir>/miniconda3/envs/test_sqm/SqueezeMeta/utils/install_utils/make_databases.pl /download/path/
(To download the latest source data and compile the databases in your server)
* perl <your home dir>/miniconda3/envs/test_sqm/SqueezeMeta/utils/install_utils/download_databases.pl /download/path/
(To download a pre-compiled version of the database, which is much quicker)
If the databases are already present in your server, you can configure this installation of SqueezeMeta with:
* perl <your home dir>/miniconda3/envs/test_sqm/SqueezeMeta/utils/install_utils/configure_nodb.pl /path/to/db
(Where /path/to/db is the path to the "db" folder generated when downloading the databases)
------------------------------------------------------------------------------
WARNING: Some SqueezeMeta dependencies could not be found in your environment or failed to execute!
- Databases were not installed, or are not configured in this installation of SqueezeMeta
提示されている選択肢としては、
- 最新のデータベースをダウンロードしてコンパイルする場合は、
make_databases.pl /download/path/を実行 - データベースのプリコンパイル済みバージョンをダウンロードするには
download_databases.pl /download/path/を実行 (←早いらしい) - データベースがすでにPCに存在する場合には、
configure_nodb.pl /path/to/dbを実行する
なのですが、圧縮済みのデータベースのtarファイルで200GBくらい、解凍すると460GBあります。
ダウンロードはSqueezeMetaの公式マニュアルに従ってダウンロードするのもいいですが、私はaria2cを使ってダウンロードしました。
また、ダウンロード場所もデータベース容量とアクセス速度を考えて、少し大きめの容量のM.2 SSDにダウンロードしました。
私がダウンロード時はSqueezeMetaが使用するDBが以下のURL下に置いてありました。
aria2cで圧縮状態のデータベースファイルをダウンロードします。
aria2c -x 10 -ctrue http://silvani.cnb.csic.es/SqueezeMeta//SqueezeMetaDB.tar.gz
一晩待てばダウンロードは終わってると思います。
解凍にはrapidgzipを使います。
インストールして実行。
mamba install -c mxmlnkn rapidgzip -y
rapidgzip -P 32 SqueezeMetaDB.tar.gz
dbという460GBくらいのフォルダが得られればデータベースの用意は終了です。
配列データの準備
以前、hostileでhost genomeを除去したデータを使います。
サンプルの詳細は以下のURLを参照してください。
- SRR18585102: Illumina shot read (Stool)
- SRR18585191: Illumina shot read (Oral)
- SRR18585213: Illumina shot read (Oral)
以前の記事で出力したSRRXXX_paired_[1|2].clean_[1|2].fastq.gzがホスト除去済みのファイルになります。
これを00fqというフォルダに配置しておきます。
mkdir 00fq
# 上記フォルダにホスト除去済みのfastqファイルをコピーしておく。
fastqファイルが用意できたらfastqファイルの情報が記載されたsampleファイルを用意します。
フォーマットはタブ区切りのヘッダー無しで、<Sample>タブ<filename>タブ<pair1|pair2>となっていればOK。sample_list.tsvという名前でファイルを作成します。
SRR18585102 SRR18585102_paired_1.clean_1.fastq.gz pair1
SRR18585102 SRR18585102_paired_2.clean_2.fastq.gz pair2
SRR18585191 SRR18585191_paired_1.clean_1.fastq.gz pair1
SRR18585191 SRR18585191_paired_2.clean_2.fastq.gz pair2
SRR18585213 SRR18585213_paired_1.clean_1.fastq.gz pair1
SRR18585213 SRR18585213_paired_2.clean_2.fastq.gz pair2
これで実行する準備は整いました。
いくつかのパイプライン実行方法
SqueezeMetaには4つの実行方法があります。
| mode | Detail |
|---|---|
| Sequential mode | 全サンプルは個別処理されて、順次分析 |
| Coassembly mode | 全サンプルのリードがプールされ、単一のアセンブリーが実行。 その後、各サンプルのリードをマッピングして、各サンプルの遺伝子量を求める。 Binningでゲノムビンを取得可能。 |
| Merged mode | サンプルを個別にAssembleして、得られたContigが1回のco-assemblyでマージされる。以降の解析はCoassemblyモードと同様。 キメラコンティグを作る可能性が高いのでCoassemblyが不可能な場合にのみ使用する。 |
| Seqmerge mode | Merged modeより多くのサンプルを扱うことが可能。ここのアセンブリーを一度にMergeせず逐次的に処理。 1. Merged modeと同様に個別アセンブリ 2.CompareMで類似度を算出し、最も類似した2つのアセンブリーをマージ 3.n個のcontigに対して、n-1回のマージステップを実行してすべてのcontigが1つにマージされるまで繰り返す |
著者のおすすめはSequential modeかCoassembly modeだが、計算リソースが制限されている場合には、Merged modeかSeqmerge modeは大体手段として使用する。その場合には、contiglenを1000以上に設定すると実行するといいらしい。
デフォルトで使用可能なAssemblerは4つ。
| Assemabler | Detail |
|---|---|
| MEGAHIT | Short read (Default), 低メモリ |
| SPAdes | Short read, アセンブリ結果が若干MEGAHITより優れている |
| Canu | Long read |
| Fly | Long read |
実行
SqueezeMeta piplineを実行します。
実行フレーズ : SqueezeMeta.pl -m <mode> -p <projectname> -s <equivfile> -f <raw fastq dir> <options>
引数 必須パラメータ
| Option | Argument |
|---|---|
| -m | sequencial, coassembly, merged, seqmerge> |
| -p | プロジェクト名 (コアセンブリおよびマージ モードでは必須) |
| -s/-samples | サンプル ファイルパス (必須) |
| -f/-seq | Fastqファイルのディレクトリ (必須) |
SqueezeMeta.pl -m coassembly -p squeeze_test -f 01fq -s sample_list.tsv sample_list.tsv -contiglen 1000 -t 32 --lowmem 100 --nokegg
実行して1晩待ったら終わってました。
確認
tree -L 2
.
├── 01fq
│ ├── SRR18585102_paired_1.clean_1.fastq.gz
│ ├── SRR18585102_paired_2.clean_2.fastq.gz
│ ├── SRR18585191_paired_1.clean_1.fastq.gz
│ ├── SRR18585191_paired_2.clean_2.fastq.gz
│ ├── SRR18585213_paired_1.clean_1.fastq.gz
│ └── SRR18585213_paired_2.clean_2.fastq.gz
├── sample_list.tsv
└── squeeze_test
├── creator.txt
├── data
├── ext_tables
├── intermediate
├── methods.txt
├── parameters.pl
├── progress
├── results
├── SqueezeMeta_conf.pl
├── syslog
└── temp
8 directories, 13 files
いい感じに全工程実行されていそうです。
結果の確認
readのmapping率の確認
- squeeze_test/results/10.squeeze_test.mappingstat
アッセンブリに関連する情報が記載されているファイルです。
Contig配列にMappingしているので、各サンプルのReadがどの程度解析に使用されたのかを確認することができます。
例えば、Mapping%が50%未満であれば、大半がアセンブルに失敗しているためリードがほとんど解析は使用されていないことを意味しています。ただこれは、サンプルのマイクロバイオーム非常に多様であったり、シーケンスの取得データ量が不十分な場合に発生する可能性があります。
そういうときは、アセンブル前のReadを使ってKraken2やMetaphlan4, Kaijuで系統解析を並行的に実施することが推奨されます。
最近のショートリードメタゲノムアセンブリの系統解析ツールの比較はこちらが参考になるかと思います。
cat squeeze_test/results/10.squeeze_test.mappingstat
| # Sample | Total reads | Mapped reads | Mapping perc | Total bases |
|---|---|---|---|---|
| SRR18585102 | 21750766 | 20134978 | 92.57 | 3072411635 |
| SRR18585191 | 15985452 | 13202505 | 82.59 | 2267264342 |
| SRR18585213 | 3489324 | 2410852 | 69.09 | 495944810 |
今回の結果だと、一番下のSRR18585213(Oral)のサンプルがmap率が低い傾向にありました。
オープンリーディングフレームの情報をまとめたテーブルデータ
- squeeze_test/results/13.squeeze_test.orftable
原核生物において原則タンパク質に翻訳される開始コドンから終始コドンの間のDNA配列領域を示す(※1)オープンリーディングフレームに関するサマリーテーブルです。
DIAMONDでアノテーションされたCOG(Clusters of Orthologous Genes)データベースの情報や、HMMER3でアノテーションされたpfamデータベースの情報の他、TPM、Read count, Coverageといった基本的な情報がひとまとめになっているので、SqueezeMetaの解析結果一覧ファイルといったところでしょうか。
※1: https://www.med.osaka-cu.ac.jp/bacteriology/b-online/omics/yougo/cds.shtml
SqueezeMeta実行のstatsファイル
- squeeze_test/results/21.squeeze_test.stats
Assambly結果や、検出されたORF,Binの数、CheckMで算出されたコンタミ率などがまとめられています。
# Parameters
Assembler: megahit
Min length of contigs: 1000
Max evalue for Diamond search:
Min identity for Diamond COG/KEGG search:
#------------------- Statistics on reads
# Assembly SRR18585102 SRR18585191 SRR18585213
Number of reads 41225542 21750766 15985452 3489324
Number of bases 5835620787 3072411635 2267264342 495944810
#------------------- Statistics on contigs
# Assembly
Number of contigs 47943
Total length 154684962
Longest contig 707200
Shortest contig 1000
N50 4740
N90 1268
Contigs at superkingdom (k) rank 47008 (98.0%), in 2 superkingdoms
Contigs at phylum (p) rank 46735 (97.5%), in 10 phyla
Contigs at class (c) rank 45753 (95.4%), in 16 classes
Contigs at order (o) rank 44793 (93.4%), in 27 orders
Contigs at family (f) rank 35499 (74.0%), in 43 families
Contigs at genus (g) rank 31192 (65.1%), in 100 genera
Contigs at species (s) rank 7246 (15.1%), in 199 species
Congruent 47712 (99.5%)
Disparity >0 232 (0.5%)
Disparity >= 0.25 147 (0.3%)
Most abundant taxa SRR18585102 SRR18585191 SRR18585213
superkingdom Bacteria Bacteria Bacteria
phylum Bacteroidota Pseudomonadota Bacillota
class Bacteroidia Betaproteobacteria Bacilli
order Bacteroidales Neisseriales Lactobacillales
family Bacteroidaceae Neisseriaceae Streptococcaceae
genus Bacteroides Neisseria Streptococcus
species Bacteroides fragilis Caudoviricetes sp. Caudoviricetes sp.
#------------------- Statistics on ORFs
# Assembly SRR18585102 SRR18585191 SRR18585213
Number of ORFs 171896 102430 79806 70561
Number of rRNAs 224 181 212 197
Number of tRNAs/tmRNAs 1323 854 803 711
ORFs by Aragorn 1323 854 803 711
ORFs by Prodigal 170349 101395 78791 69653
ORFs by barrnap 224 181 212 197
Orphans (no hits) 4145 2307 2221 1857
No tax assigned (with hits) 4413 2690 1443 1114
COG annotations 146525 89381 68721 60460
Pfam annotations 110860 69410 52130 46893
#------------------- Statistics on bins
# DAS
Number of bins 60
Complete >= 50% 29
Complete >= 75% 18
Complete >= 90% 13
Contamination < 10% 47
Contamination >= 50% 3
Congruent bins 17
Disparity >0 43
Disparity >= 0.25 18
Hi-qual bins (>90% complete,<10% contam) 8
Good-qual bins (>75% complete,<10% contam) 11
SqueezeMetaの結果を使った下流分析 (anvi'o)
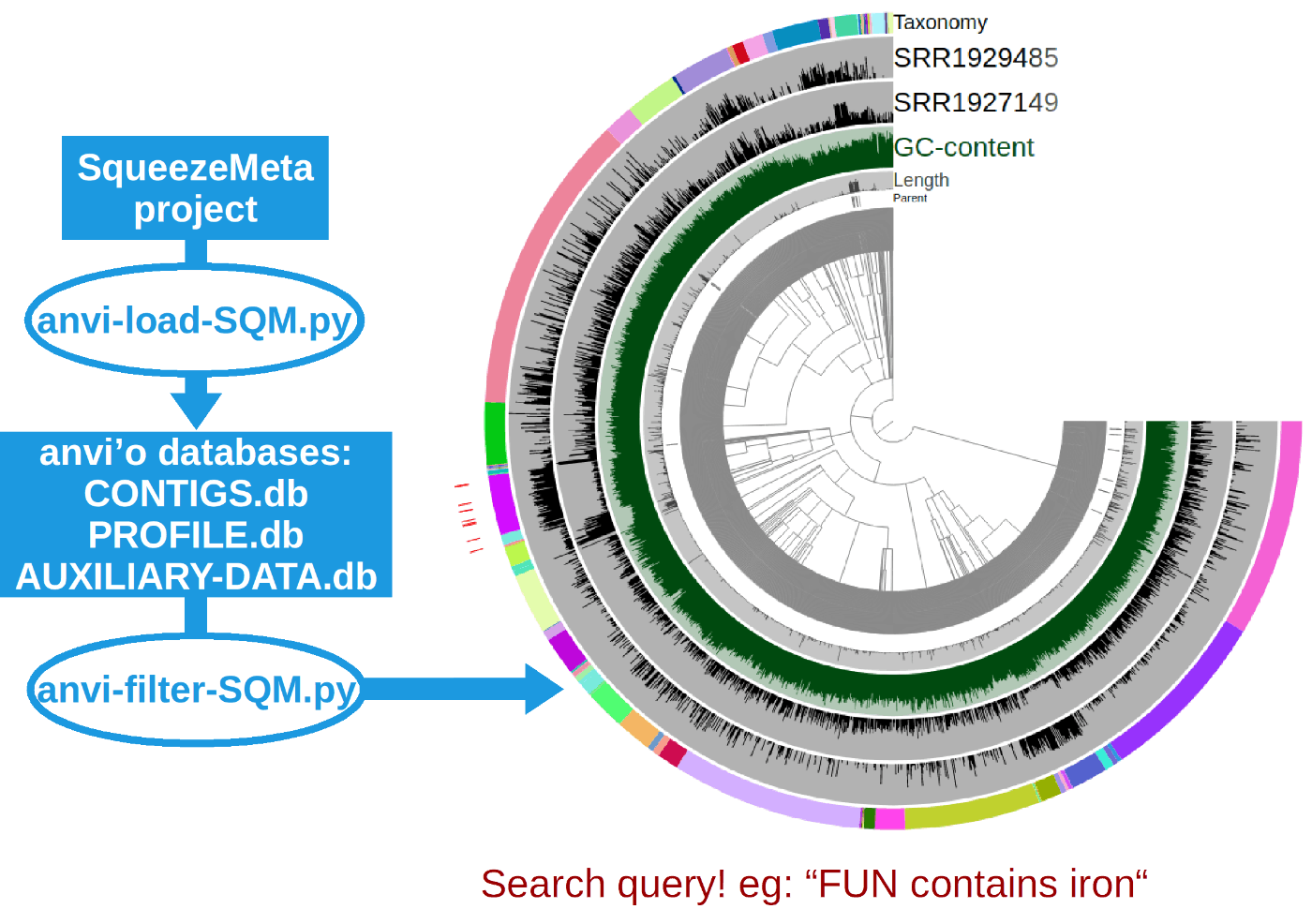
以下のwikiに従って進めます。
1. anvi'oのインストールと仮想環境の有効化
最新版のanvi'o v8はエラーが出ます。SQM-toolのサポートはanvio 6.2のようですが、v7をインストールします。
# 仮想環境の作成
mamba create -y -n anvio-7 python=3.6
# acativate
conda activate anvio-7
# 依存関係のインストール
mamba install prodigal hmmer dendropy
# githubから過去のバージョンのAnvi'oをダウンロード
curl -L https://github.com/merenlab/anvio/releases/download/v7.1/anvio-7.1.tar.gz --output anvio-7.1.tar.gz
# pipでインストール
pip install anvio-7.1
# test run
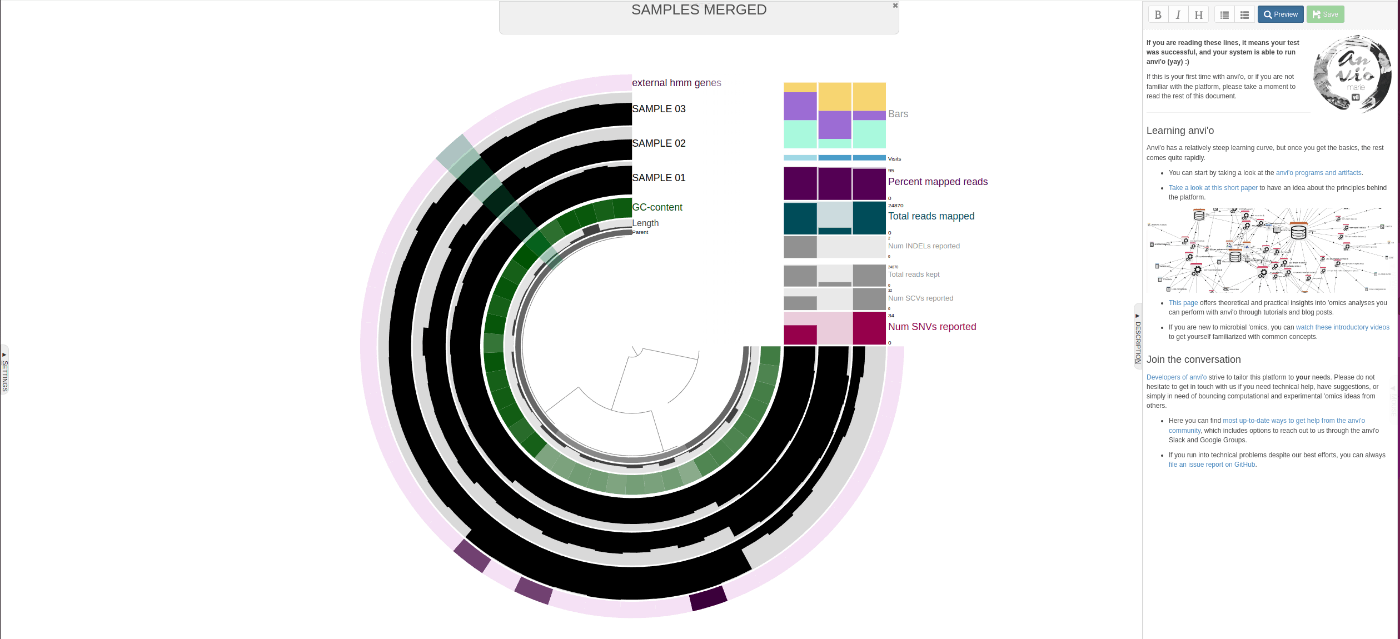
anvi-self-test --suite mini
時々、warningが出ますが、ブラウザが立ち上がって以下のような図が出ればOK。
anvi-load-sqm.pyを実行して、anvi'o用のデータを生成
ではSqueezeMetaの結果をanvi'o用に整形していきます。
まずは実行ファイルのパスの確認。検索に結構時間がかかるようでしたら、多分、対象スクリプトは
<your home dir>/miniconda3/envs/SqueezeMeta/utils/anvio_utils/にあるので見てみてください。
find . -type f -name "anvi-load-sqm.py"
上記編集ができれば実行準備完了。実行します。
python3 <your home dir>/miniconda3/envs/SqueezeMeta/SqueezeMeta/utils/anvio_utils/anvi-load-sqm.py \
-p squeeze_test \
-o squeeze_test/results/anvio \
--num-threads 32 \
--min-contig-length 10000 \
--run-scg-taxonomy \
--profile-SCVs \
--run-HMMS
問題なく完了すると、-oで指定したフォルダにCONTIGS.dbとPROFILE.dbが出力されます。
ls squeeze_test/results/anvio/*.db -1
squeeze_test/results/anvio/AUXILIARY-DATA.db
squeeze_test/results/anvio/CONTIGS.db
squeeze_test/results/anvio/PROFILE.db
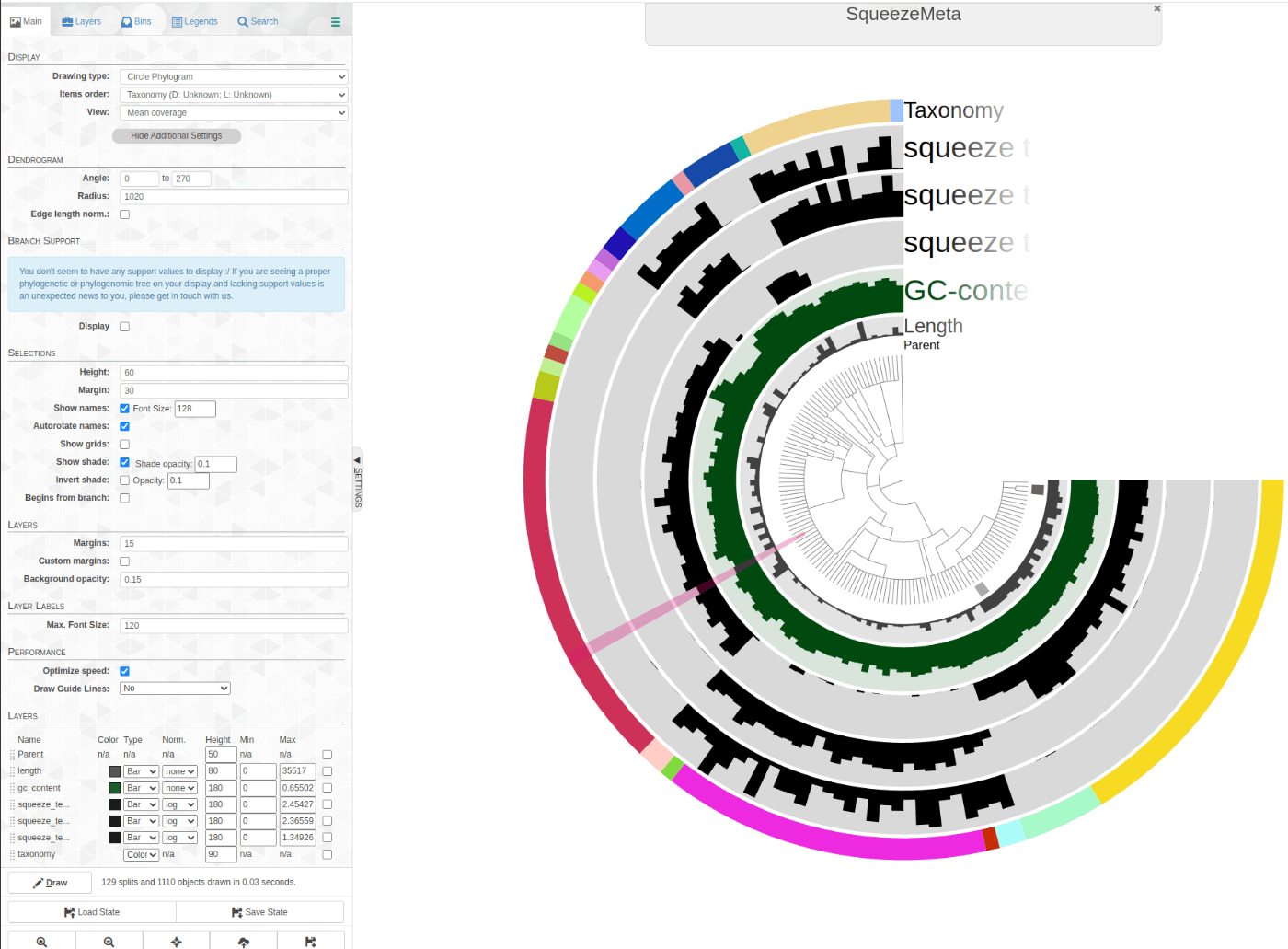
anvi-filter-sqm.pyで情報をフィルタリングして視覚化する
Contig数が多い場合には、フィルタリングすることで図がスッキリします。
Case1. antibiotic (抗生物質)とannotationのついた遺伝子を含むcontigを視覚化
python3 <your home dir>/miniconda3/envs/SqueezeMeta/SqueezeMeta/utils/anvio_utils/anvi-filter-sqm.py \
-c squeeze_test/results/anvio/CONTIGS.db \
-p squeeze_test/results/anvio/PROFILE.db \
-t squeeze_test/results/anvio/squeeze_test_anvio_contig_taxonomy.txt \
-o squeeze_test/results/anvio/antibiotic \
-q '(FUN CONTAINS antibiotic)'
ブラウザが立ち上がったらDrawing typeなどを選択して、左下のDrawをクリックするとグラフが表示されます。
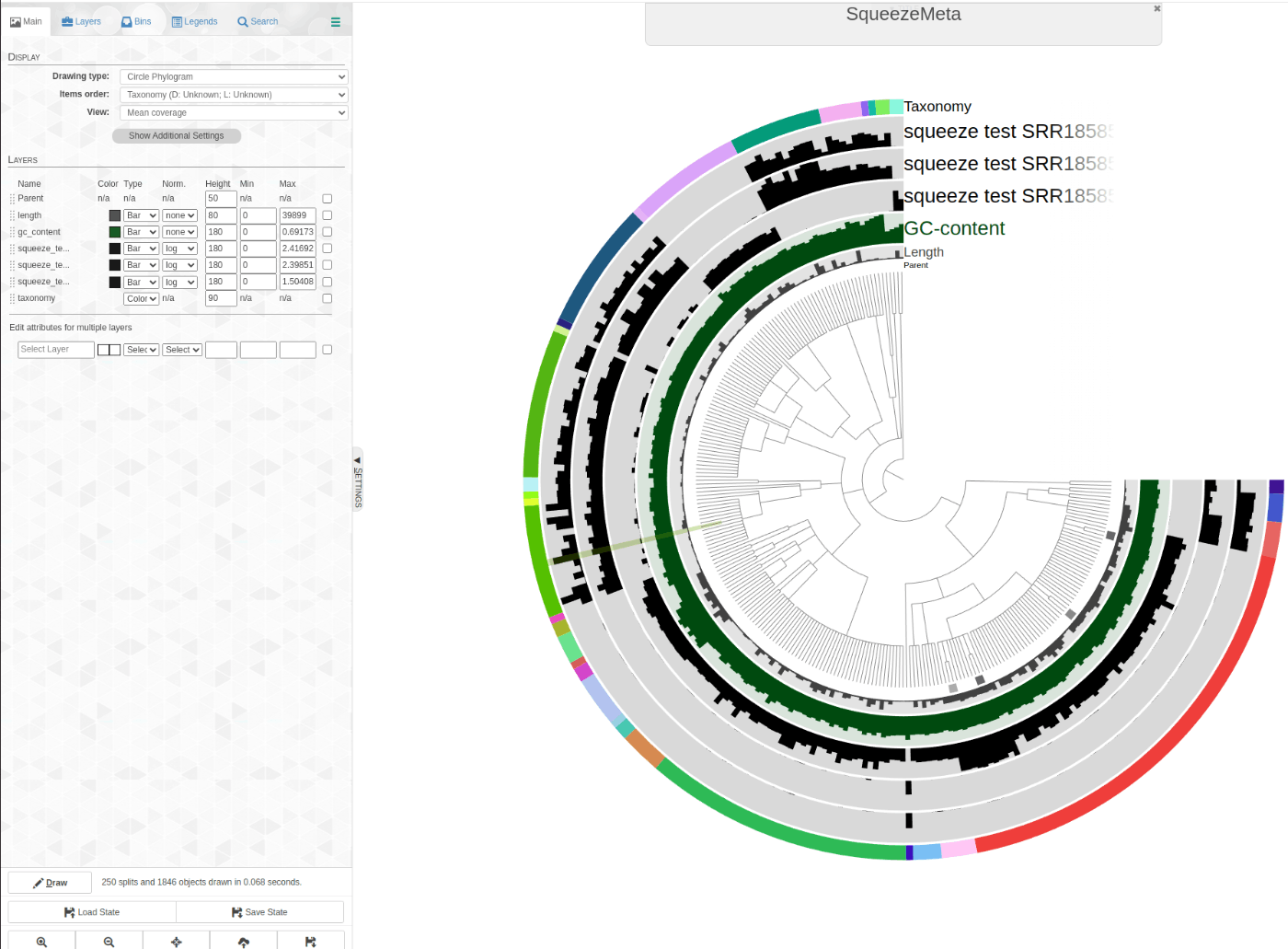
Case2. aromatic (芳香)の経路に関連する遺伝子を含むcontigを可視化
python3 <your home dir>/miniconda3/envs/SqueezeMeta/SqueezeMeta/utils/anvio_utils/anvi-filter-sqm.py \
-c squeeze_test/results/anvio/CONTIGS.db \
-p squeeze_test/results/anvio/PROFILE.db \
-t squeeze_test/results/anvio/squeeze_test_anvio_contig_taxonomy.txt \
-o squeeze_test/results/anvio/aromatic \
-q '(FUN CONTAINS aromatic)'
Case3. anvi'o を使用してビンを探索する
Binの状況を確認
anvi-show-collections-and-bins -p squeeze_test/results/anvio/PROFILE.db
SqueezMetaだとDAS_Toolsで複数のBinnerの結果をまとめているので、DASというコレクションが1つあるはずです。
すべてのBinの情報を使って表示。
anvi-interactive -c squeeze_test/results/anvio/CONTIGS.db -p squeeze_test/results/anvio/PROFILE.db -C DAS
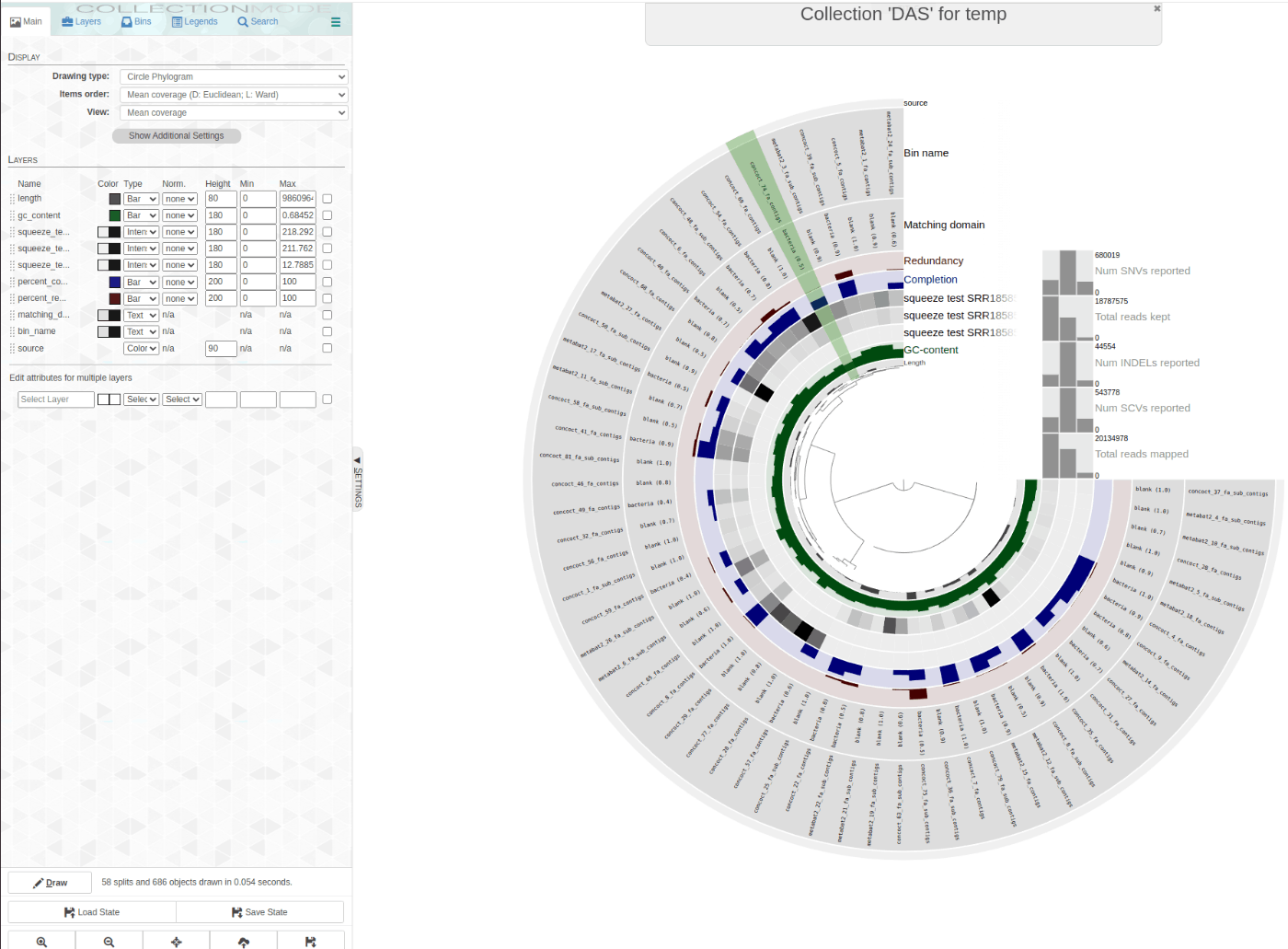
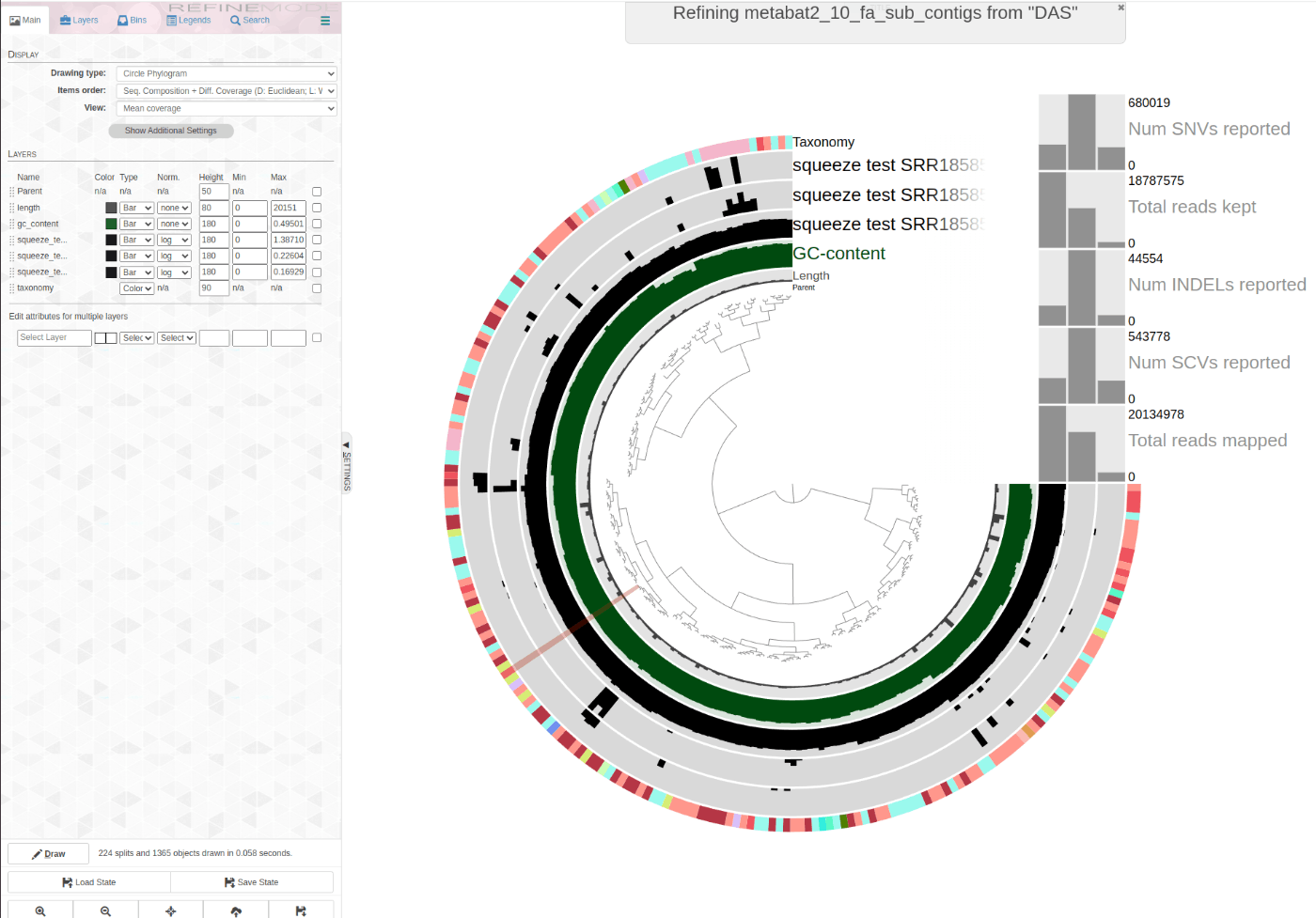
特定のBinに着目して表示。
anvi-refine -c squeeze_test/results/anvio/CONTIGS.db -p squeeze_test/results/anvio/PROFILE.db -C DAS -b metabat2_10_fa_sub_contigs
今回はこれでおしまい。
Discussion