hostile: 細菌のショットガンメタゲノム Host genome除去
腸内や口腔の細菌研究で利用される、All in Oneのホストゲノム除去ツールとしては、Kneaddataが有名所です。
一方、T2Tのリファレンス配列を使い精度を上げつつ、比較的低いPCスペックの要件でホストとなるヒトゲノムを除去するhostileが最近開発されました。
ホストゲノムの除去ステップにおいて、GRCh38よりも配列網羅性の高いT2T-CHM13を使用することは理にかなっていると思います。
デフォルトでT2Tを採用しているhostileとKneaddataでツールとしてどんな違いがあるのか、口腔細菌と腸内細菌の配列データに対して適用して比較してみます。
ツールのインストール
mamba create -n rm_host_env hostile kneaddata seqkit -y
mamba activate rm_host_env
keanddata用にTrimmomaticはReleasesページのtar.gzファイルをダウンロード&解答して用意しておく。
wget https://github.com/usadellab/Trimmomatic/files/5854859/Trimmomatic-0.39.zip
unzip Trimmomatic-0.39.zip
rm Trimmomatic-0.39.tar.gz
# 確認
java -jar Trimmomatic-0.39/trimmomatic-0.39.jar -h
処理の流れ
Illumina short readの処理Flowです。
※ KneaddataでTRFは--trf <path to trf>で使用できるオプション扱いでした。
hostile
USAGE
usage: hostile [-h] [--version] {clean,mask,fetch} ...
positional arguments:
{clean,mask,fetch}
clean Remove reads aligning to a target genome from fastq[.gz] input files
mask Mask reference genome against target genome(s)
fetch Download reference genomes (Minimap2) and/or indexes (Bowtie2). Run without
arguments to fetch defaults
options:
-h, --help show this help message and exit
--version show program's version number and exit
利用可能なデータベースの情報を確認
hostile fetch --list-available
22:26:31 INFO: HTTP Request: GET https://objectstorage.uk-london-1.oraclecloud.com/n/lrbvkel2wjot/b/human-genome-bucket/o "HTTP/1.1 200 OK"
hla-v3.51.0.fa.gz (Minimap2)
human-t2t-hla-argos985-mycob140.fa.gz (Minimap2)
human-t2t-hla-argos985-mycob140.tar (Bowtie2)
human-t2t-hla-argos985.fa.gz (Minimap2)
human-t2t-hla-argos985.tar (Bowtie2)
human-t2t-hla.fa.gz (Minimap2; DEFAULT)
human-t2t-hla.tar (Bowtie2; DEFAULT)
mit-nc_012920.1.fa.gz (Minimap2)
t2t-chm13v2.0.fa.gz (Minimap2)
デフォルトのReference genomeをダウンロード
hostile fetch
デフォルト以外のReference genomeをダウンロードする場合はこちら。
hostile fetch --filename human-t2t-hla-argos985-mycob140.fa.gz
配列データの準備
SRAから配列データを取得。Illuminaのライブラリ調整キットはNexteraXTです(Trimmomaticで使う情報)。
- SRR18585102: Illumina shot read (Stool)
- SRR18585191: Illumina shot read (Oral)
- SRR18585213: Illumina shot read (Oral)
以降でのツール実行のためにACCでArrayを作っときます。
illumina_fq_array=("SRR18585102" "SRR18585191" "SRR18585213")
for illumina in ${illumina_fq_array[@]}; do
aria2c -x10 -ctrue https://sra-pub-run-odp.s3.amazonaws.com/sra/${illumina}/${illumina}
done
for illumina in ${illumina_fq_array[@]}; do
fasterq-dump -S -p -e 32 ${illumina}
done
ダウンロードしたデータは以下のような感じです。
mkdir 02Stats
seqkit stats -a -T 00fq/*fastq.gz > 02Stats/Rawfq_stats.tsv
| file | num_seqs (M read) | sum_len (Gbp) | Q30(%) |
|---|---|---|---|
| SRR18585102_1.fastq.gz | 12 | 1.7 | 87.36 |
| SRR18585102_2.fastq.gz | 12 | 1.7 | 91.90 |
| SRR18585191_1.fastq.gz | 10 | 1.4 | 89.96 |
| SRR18585191_2.fastq.gz | 10 | 1.4 | 87.41 |
| SRR18585213_1.fastq.gz | 4.6 | 0.7 | 88.26 |
| SRR18585213_2.fastq.gz | 4.6 | 0.7 | 82.49 |
kneaddataの実行
Kneaddataは実行すれば出力として、アダプタートリミングとホスト除去をしたFastqファイルを出力します。
mkdir 01kneaddata
fq_path="00fq"
illumina_fq_array=("SRR18585102" "SRR18585191"
"SRR18585213")
for illumina in ${illumina_fq_array[@]};do
kneaddata \
--trimmomatic ~/Desktop/bioinfo/host_remove/Trimmomatic-0.39/ \
-t 32 \
-i1 "${fq_path}/${illumina}_1.fastq.gz" \
-i2 "${fq_path}/${illumina}_2.fastq.gz" \
--reference-db kneaddata_human_genome_bowtie2_reference_db \
--output-prefix "${illumina}" \
--log "${illumina}"_keanddata.log \
--trimmomatic-options "ILLUMINACLIP:$PWD/Trimmomatic-0.39/adapters/NexteraPE-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50" \
-o "01kneaddata/${illumina}";
pigz -p 24 "01kneaddata/${illumina}/*.fastq"
done
興味があったので、kneaddataにT2Tのデータも使ってみます。まず、bowtie2用のデータベースをビルドします。リファレンスファイルはhostileでダウンロードされたのものを使用しています。
bowtie2-build -f human-t2t-hla.fa.gz human-t2t-hla
データベースのビルドは結構時間かかりました。
for illumina in ${illumina_fq_array[@]}; do
kneaddata \
--trimmomatic ~/Desktop/bioinfo/host_remove/Trimmomatic-0.39/ \
-t 32 \
-i1 "${fq_path}/${illumina}_1.fastq.gz" \
-i2 "${fq_path}/${illumina}_2.fastq.gz" \
--reference-db human-t2t-hla \
--output-prefix "${illumina}_t2t" \
--log "${illumina}_t2t_keanddata.log" \
--trimmomatic-options "ILLUMINACLIP:$PWD/Trimmomatic-0.39/adapters/NexteraPE-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50" \
-o "01kneaddata/${illumina}_t2t"
done
pigz -p 24 01kneaddata/*/*.fastq
hostileの実行
1. Trimmomaticによるアダプタートリミング
hostileはホストゲノムの除去だけなので、ショートリードについてはKneaddataと同様、まずTrimmomaticでアダプタートリミングします。
トリミングとフィルタリングに関するパラメーターはKeaddataで使用されていたものを指定します。
input_path="00fq"
hostile_path="01hostile"
mkdir ${hostile_path}
illumina_fq_array=("SRR18585102" "SRR18585191" "SRR18585213")
for illumina in ${illumina_fq_array[@]};do
java -jar $PWD/Trimmomatic-0.39/trimmomatic-0.39.jar \
PE \
-trimlog ${illumina}_trimmomatic.log \
-threads 32 \
${input_path}/${illumina}_1.fastq.gz \
${input_path}/${illumina}_2.fastq.gz \
${hostile_path}/${illumina}_paired_1.fastq.gz \
${hostile_path}/${illumina}_unpaired_1.fastq.gz \
${hostile_path}/${illumina}_paired_2.fastq.gz \
${hostile_path}/${illumina}_unpaired_2.fastq.gz \
ILLUMINACLIP:$PWD/Trimmomatic-0.39/adapters/NexteraPE-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50
done
2.fastaファイルへの変換
TRF用にアダプタートリミング済みのFastqファイルをfastaファイルに変換する
hostile_path="01hostile"
illumina_fq_array=("SRR18585102" "SRR18585191" "SRR18585213")
for illumina in ${illumina_fq_array[@]};do
seqkit fq2fa ${hostile_path}/${illumina}_paired_1.fastq.gz \
--out-file ${hostile_path}/${illumina}_paired_1.fasta
done
for illumina in ${illumina_fq_array[@]};do
seqkit fq2fa ${hostile_path}/${illumina}_paired_2.fastq.gz \
--out-file ${hostile_path}/${illumina}_paired_2.fasta
done
3. TRFによるタンデムリピート配列の検出
次に、Tandem Repeat finderでタンデムリピートを検出する
まずはインストール
mamba install bioconda::trf
trfもkneaddataと同じ設定で行きます。
kneaddataのtrf argunemt:
trf arguments:
--trf TRF_PATH path to TRF
[ DEFAULT : $PATH ]
--match MATCH matching weight
[ DEFAULT : 2 ]
--mismatch MISMATCH mismatching penalty
[ DEFAULT : 7 ]
--delta DELTA indel penalty
[ DEFAULT : 7 ]
--pm PM match probability
[ DEFAULT : 80 ]
--pi PI indel probability
[ DEFAULT : 10 ]
--minscore MINSCORE minimum alignment score to report
[ DEFAULT : 50 ]
--maxperiod MAXPERIOD
maximum period size to report
[ DEFAULT : 500 ]
上記を参考にtrfを実行していきます。
seqkit fq2faで作成したfastaファイルをinputとして、trfでタンデムリピート配列の検出を実施します。kneaddata内で行われているようなparallel処理をしていないので少し時間がかかります。
hostile_path="01hostile"
trf_match=2
trf_mismatch=7
trf_delta=7
trf_match_probability=80
trf_pi=10
trf_minscore=50
trf_maxperiod=500
# R1
for illumina in ${illumina_fq_array[@]};do
trf \
${hostile_path}/${illumina}_paired_1.fasta \
${trf_match} \
${trf_mismatch} \
${trf_delta} \
${trf_match_probability} \
${trf_pi} \
${trf_minscore} \
${trf_maxperiod} \
-ngs \
-h > ${hostile_path}/${illumina}_paired_1.repeatout
done
# R2
for illumina in ${illumina_fq_array[@]};do
trf \
${hostile_path}/${illumina}_paired_2.fasta \
${trf_match} \
${trf_mismatch} \
${trf_delta} \
${trf_match_probability} \
${trf_pi} \
${trf_minscore} \
${trf_maxperiod} \
-ngs \
-h > ${hostile_path}/${illumina}_paired_2.repeatout
done
01hostileフォルダ内に.repeatoutフォルダが生成されていたらOKです。
ここまできたら、fastaファイルは削除するか圧縮しておきましょう
hostile_path="01hostile"
pigz -p 32 ${hostile_path}/*.fasta
4.seqkitによるタンデムリピート配列の除去
trfでタンデムリピートが検出された配列をseqkit grepでRaw fastqファイルから除去します。
hostile_path="01hostile"
illumina_fq_array=("SRR18585102" "SRR18585191" "SRR18585213")
for illumina in ${illumina_fq_array[@]};do
# R1
grep "^@" ${hostile_path}/${illumina}_paired_1.repeatout | \
sed 's/@//'> ${hostile_path}/${illumina}_paired_1.repeatseq_list
# R2
grep "^@" ${hostile_path}/${illumina}_paired_2.repeatout | \
sed 's/@//'> ${hostile_path}/${illumina}_paired_2.repeatseq_list
# seqkitを使用して、対象となるシーケンスを除外
# R1
seqkit \
grep -n -v \
-f ${hostile_path}/${illumina}_paired_1.repeatseq_list \
-o ${hostile_path}/${illumina}.repeats.removed.1.fastq.gz \
${hostile_path}/${illumina}_paired_1.fastq.gz
# R1
seqkit \
grep -n -v \
-f ${hostile_path}/${illumina}_paired_2.repeatseq_list \
-o ${hostile_path}/${illumina}.repeats.removed.2.fastq.gz \
${hostile_path}/${illumina}_paired_2.fastq.gz
done
5.hostileによるホストゲノム除去
アダプタートリムとクオリティフィルタリングが実行後、タンデムリピート配列が除去された配列データに対してhostileを実行します。
hostile_path="01hostile"
# For Illumina seq data(short read)
for illumina in ${illumina_fq_array[@]};do
hostile clean \
--fastq1 ${hostile_path}/${illumina}_paired_1.fastq.gz \
--fastq2 ${hostile_path}/${illumina}_paired_2.fastq.gz \
--aligner bowtie2 \
--out-dir ${hostile_path}/${illumina} \
--threads 32
done
比較
各処理段階で生成されたfastqファイルのread数などの基本情報をseqkit statsで取得します。
-Tオプションをつけることでタブ区切り出力ができます。
mkdir 02Stats
# hostile
## After Trimming
seqkit stats -a -T 01hostile/*_paired*.fastq.gz > 02Stats/hostile_trimmed.tsv
## After TRF
seqkit stats -a -T 01hostile/*.repeats*.fastq.gz > 02Stats/hostile_repeat.tsv
## After host remove
seqkit stats -a -T 01hostile/*/*clean*.fastq.gz > 02Stats/host
ile_cleaned.tsv
# cat
cat 02Stats/*.tsv | head -n 1 > 02Stats/hostile_merged && \
find 02Stats -type f -name "hostile*.tsv" -exec tail -n +2 {} \; >> 02Stats/hostile_merged
# Kneaddata
## After Trimming
seqkit stats -a -T 01kneaddata/*/*.trimmed*.fastq.gz > 02Stats
/kneaddata_trimmed.tsv
## After TRF
seqkit stats -a -T 01kneaddata/*/*.repeats.removed*.fastq.gz > 02Stats/kneaddata_repeat.tsv
## After host remove
seqkit stats -a -T 01kneaddata/*/*_paired_*.fastq.gz > 02Stats/kneaddata_cleaned.tsv
#cat
cat 02Stats/*.tsv | head -n 1 > 02Stats/kneaddata_merged && find 02Stats -type f -name "kneaddata*.tsv" -exec tail -n +2 {} \; >> 02Stats/kneaddata_merged
hostile
awk -F'\t' -v OFS='\t' '{print $1, $4, $5, $7, $15}' 02Stats/hostile_merged
少し順番変えたりして編集していますが以下通りです。
- paired.fastq.gz : trimmomatic後
- repeats.fastq.gz: TRF後
- clean.fastq.gz : ホストゲノム削除後
- SRR18585102
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585102_paired_1.fastq.gz | 10878462 | 1520551182 | 139.8 | 90.27 |
| SRR18585102_paired_2.fastq.gz | 10878462 | 1552679667 | 142.7 | 93.88 |
| SRR18585102.repeats.removed.1.fastq.gz | 10840698 | 1515199817 | 139.8 | 90.28 |
| SRR18585102.repeats.removed.2.fastq.gz | 10836120 | 1546706791 | 142.7 | 93.89 |
| SRR18585102_paired_1.clean_1.fastq.gz | 10875383 | 1520142585 | 139.8 | 90.27 |
| SRR18585102_paired_2.clean_2.fastq.gz | 10875383 | 1552269050 | 142.7 | 93.88 |
- SRR18585191
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585191_paired_1.fastq.gz | 9001636 | 1276327170 | 141.8 | 93.19 |
| SRR18585191_paired_2.fastq.gz | 9001636 | 1262435801 | 140.2 | 91.78 |
| SRR18585191.repeats.removed.1.fastq.gz | 8769101 | 1245733846 | 142.1 | 93.27 |
| SRR18585191.repeats.removed.2.fastq.gz | 8769680 | 1233239195 | 140.6 | 91.85 |
| SRR18585191_paired_1.clean_1.fastq.gz | 7992726 | 1137493144 | 142.3 | 93.38 |
| SRR18585191_paired_2.clean_2.fastq.gz | 7992726 | 1129771198 | 141.3 | 92.29 |
- SRR18585213
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585213_paired_1.fastq.gz | 3917916 | 550644840 | 140.5 | 93.06 |
| SRR18585213_paired_2.fastq.gz | 3917916 | 534400868 | 136.4 | 89.86 |
| SRR18585213.repeats.removed.1.fastq.gz | 3614055 | 511332914 | 141.5 | 93.29 |
| SRR18585213.repeats.removed.2.fastq.gz | 3618283 | 497795747 | 137.6 | 89.99 |
| SRR18585213_paired_1.clean_1.fastq.gz | 1744662 | 248862682 | 142.6 | 93.91 |
| SRR18585213_paired_2.clean_2.fastq.gz | 1744662 | 247082128 | 141.6 | 92.63 |
Kneaddata
awk -F'\t' -v OFS='\t' '{print $1, $4, $5, $7, $15}' 02Stats/kneaddata_merged
こちらも不要なデータは除去しましたが、以下のような結果になりました。
- GRCh37: SRR18585102
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585102.trimmed.1.fastq.gz | 10878462 | 1520551182 | 139.8 | 90.27 |
| SRR18585102.trimmed.2.fastq.gz | 10878462 | 1552679667 | 142.7 | 93.88 |
| SRR18585102.repeats.removed.1.fastq.gz | 10840698 | 1515199817 | 139.8 | 90.28 |
| SRR18585102.repeats.removed.2.fastq.gz | 10836120 | 1546706791 | 142.7 | 93.89 |
| SRR18585102_paired_1.fastq.gz | 10803371 | 1510030745 | 139.8 | 90.28 |
| SRR18585102_paired_2.fastq.gz | 10803371 | 1542039446 | 142.7 | 93.89 |
- GRCh37: SRR18585191
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585191.trimmed.1.fastq.gz | 9001636 | 1276327170 | 141.8 | 93.19 |
| SRR18585191.trimmed.2.fastq.gz | 9001636 | 1262435801 | 140.2 | 91.78 |
| SRR18585191.repeats.removed.1.fastq.gz | 8769101 | 1245733846 | 142.1 | 93.27 |
| SRR18585191.repeats.removed.2.fastq.gz | 8769680 | 1233239195 | 140.6 | 91.85 |
| SRR18585191_paired_1.fastq.gz | 7908916 | 1125803021 | 142.3 | 93.39 |
| SRR18585191_paired_2.fastq.gz | 7908916 | 1118315312 | 141.4 | 92.30 |
- GRCh37: SRR18585213
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585213.trimmed.1.fastq.gz | 3917916 | 550644840 | 140.5 | 93.06 |
| SRR18585213.trimmed.2.fastq.gz | 3917916 | 534400868 | 136.4 | 89.86 |
| SRR18585213.repeats.removed.1.fastq.gz | 3614055 | 511332914 | 141.5 | 93.29 |
| SRR18585213.repeats.removed.2.fastq.gz | 3618283 | 497795747 | 137.6 | 89.99 |
| SRR18585213_paired_1.fastq.gz | 1714711 | 244771628 | 142.7 | 93.93 |
| SRR18585213_paired_2.fastq.gz | 1714711 | 243063929 | 141.8 | 92.66 |
- t2t: SRR18585102
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585102_t2t.trimmed.1.fastq.gz | 10878462 | 1520551182 | 139.8 | 90.27 |
| SRR18585102_t2t.trimmed.2.fastq.gz | 10878462 | 1552679667 | 142.7 | 93.88 |
| SRR18585102_t2t.repeats.removed.1.fastq.gz | 10840698 | 1515199817 | 139.8 | 90.28 |
| SRR18585102_t2t.repeats.removed.2.fastq.gz | 10836120 | 1546706791 | 142.7 | 93.89 |
| SRR18585102_t2t_paired_1.fastq.gz | 10803720 | 1510080395 | 139.8 | 90.28 |
| SRR18585102_t2t_paired_2.fastq.gz | 10803720 | 1542089784 | 142.7 | 93.89 |
- t2t: SRR18585191
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585191_t2t.trimmed.1.fastq.gz | 9001636 | 1276327170 | 141.8 | 93.19 |
| SRR18585191_t2t.trimmed.2.fastq.gz | 9001636 | 1262435801 | 140.2 | 91.78 |
| SRR18585191_t2t.repeats.removed.1.fastq.gz | 8769101 | 1245733846 | 142.1 | 93.27 |
| SRR18585191_t2t.repeats.removed.2.fastq.gz | 8769680 | 1233239195 | 140.6 | 91.85 |
| SRR18585191_t2t_paired_1.fastq.gz | 7909139 | 1125836068 | 142.3 | 93.39 |
| SRR18585191_t2t_paired_2.fastq.gz | 7909139 | 1118346860 | 141.4 | 92.30 |
- t2t: SRR18585213
| file | num_seqs | sum_len | avg_len | Q30(%) |
|---|---|---|---|---|
| SRR18585213_t2t.trimmed.1.fastq.gz | 3917916 | 550644840 | 140.5 | 93.06 |
| SRR18585213_t2t.trimmed.2.fastq.gz | 3917916 | 534400868 | 136.4 | 89.86 |
| SRR18585213_t2t.repeats.removed.1.fastq.gz | 3614055 | 511332914 | 141.5 | 93.29 |
| SRR18585213_t2t.repeats.removed.2.fastq.gz | 3618283 | 497795747 | 137.6 | 89.99 |
| SRR18585213_t2t_paired_1.fastq.gz | 1714989 | 244812533 | 142.7 | 93.93 |
| SRR18585213_t2t_paired_2.fastq.gz | 1714989 | 243103755 | 141.8 | 92.65 |
処理ごとのRead数の推移をグラフにしてみました。使用したcsvデータはこちらにおいてあります。
python scripts
- hostile
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# データの読み込み
df = pd.read_csv("03plot/hostile_res.csv", sep=",")
df['dataset'] = df['file'].str.extract('(SRR\d+)')
df['stage'] = np.where(df['file'].str.contains('clean'), 'Cleaned',
np.where(df['file'].str.contains('repeats.removed'), 'Repeats Removed',
np.where(df['file'].str.contains('paired'), 'Trimmed', 'Other')))
df['sub_dataset'] = df['file'].str.extract('([1|2].fastq.gz)')
df = df.sort_values('stage', key=lambda col: col.map({'Trimmed': 0, 'Repeats Removed': 1, 'Cleaned': 2})).reset_index(drop=True)
sums_by_dataset_stage = df.groupby(['dataset', 'sub_dataset', 'stage'])['num_seqs'].sum().unstack()
# グラフの描画
fig, ax = plt.subplots(figsize=(15, 6))
width = 0.2 # 棒グラフの幅
dataset_stages = sums_by_dataset_stage.index
x = np.arange(len(dataset_stages)) # x軸の位置
# 各データセットと処理段階の組み合わせごとに棒グラフを描画
for i, stage in enumerate(['Trimmed', 'Repeats Removed', 'Cleaned']):
if stage in sums_by_dataset_stage.columns:
ax.bar(x + i * width, sums_by_dataset_stage[stage], width, label=stage)
# 軸ラベル、タイトル、凡例の設定
ax.set_ylabel('Number of Sequences')
ax.set_title('Number of Sequences at Each Processing Stage : hostile')
ax.set_xticks(x + width)
ax.set_xticklabels([f'{ds[0]} {ds[1]}' for ds in dataset_stages], rotation=45)
ax.legend(title="Processing Stages")
plt.show()
- Kneaddata: CRCh37
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# データの読み込み
df = pd.read_csv("03plot/knead_grch37_res.csv", sep=",")
# 各データセットごとに棒グラフを作成するためのスクリプト
# データフレームにデータセットの識別子を追加
df['dataset'] = df['file'].str.extract('(SRR\d+)')
df['stage'] = np.where(df['file'].str.contains('paired'), 'Cleaned',
np.where(df['file'].str.contains('repeats.removed'), 'Repeats Removed',
np.where(df['file'].str.contains('trimmed'), 'Trimmed', 'Other')))
# データセットを細分化するための識別子を追加
df['sub_dataset'] = df['file'].str.extract('([1|2].fastq.gz)')
df = df.sort_values('stage', key=lambda col: col.map({'Trimmed': 0, 'Repeats Removed': 1, 'Cleaned': 2})).reset_index(drop=True)
# 各データセットと処理段階ごとにシーケンス数の合計を計算
sums_by_dataset_stage = df.groupby(['dataset', 'sub_dataset', 'stage'])['num_seqs'].sum().unstack()
# グラフの描画
fig, ax = plt.subplots(figsize=(15, 6))
width = 0.2 # 棒グラフの幅
dataset_stages = sums_by_dataset_stage.index
x = np.arange(len(dataset_stages)) # x軸の位置
# 各データセットと処理段階の組み合わせごとに棒グラフを描画
for i, stage in enumerate(['Trimmed', 'Repeats Removed', 'Cleaned']):
if stage in sums_by_dataset_stage.columns:
ax.bar(x + i * width, sums_by_dataset_stage[stage], width, label=stage)
# 軸ラベル、タイトル、凡例の設定
ax.set_ylabel('Number of Sequences')
ax.set_title('Number of Sequences at Each Processing Stage : kneaddata_GRCh37')
ax.set_xticks(x + width)
ax.set_xticklabels([f'{ds[0]} {ds[1]}' for ds in dataset_stages], rotation=45)
ax.legend(title="Processing Stages")
plt.show()
- Kneaddata: t2t
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# データの読み込み
df = pd.read_csv("03plot/knead_t2t_res.csv", sep=",")
# 各データセットごとに棒グラフを作成するためのスクリプト
# データフレームにデータセットの識別子を追加
df['dataset'] = df['file'].str.extract('(SRR\d+)')
df['stage'] = np.where(df['file'].str.contains('paired'), 'Cleaned',
np.where(df['file'].str.contains('repeats.removed'), 'Repeats Removed',
np.where(df['file'].str.contains('trimmed'), 'Trimmed', 'Other')))
# データセットを細分化するための識別子を追加
df['sub_dataset'] = df['file'].str.extract('([1|2].fastq.gz)')
df = df.sort_values('stage', key=lambda col: col.map({'Trimmed': 0, 'Repeats Removed': 1, 'Cleaned': 2})).reset_index(drop=True)
# 各データセットと処理段階ごとにシーケンス数の合計を計算
sums_by_dataset_stage = df.groupby(['dataset', 'sub_dataset', 'stage'])['num_seqs'].sum().unstack()
# グラフの描画
fig, ax = plt.subplots(figsize=(15, 6))
width = 0.2 # 棒グラフの幅
dataset_stages = sums_by_dataset_stage.index
x = np.arange(len(dataset_stages)) # x軸の位置
# 各データセットと処理段階の組み合わせごとに棒グラフを描画
for i, stage in enumerate(['Trimmed', 'Repeats Removed', 'Cleaned']):
if stage in sums_by_dataset_stage.columns:
ax.bar(x + i * width, sums_by_dataset_stage[stage], width, label=stage)
# 軸ラベル、タイトル、凡例の設定
ax.set_ylabel('Number of Sequences')
ax.set_title('Number of Sequences at Each Processing Stage : kneaddata_t2t')
ax.set_xticks(x + width)
ax.set_xticklabels([f'{ds[0]} {ds[1]}' for ds in dataset_stages], rotation=45)
ax.legend(title="Processing Stages")
plt.show()
-
hostile

-
Kneaddata: CRCh37

-
Kneaddata: t2t
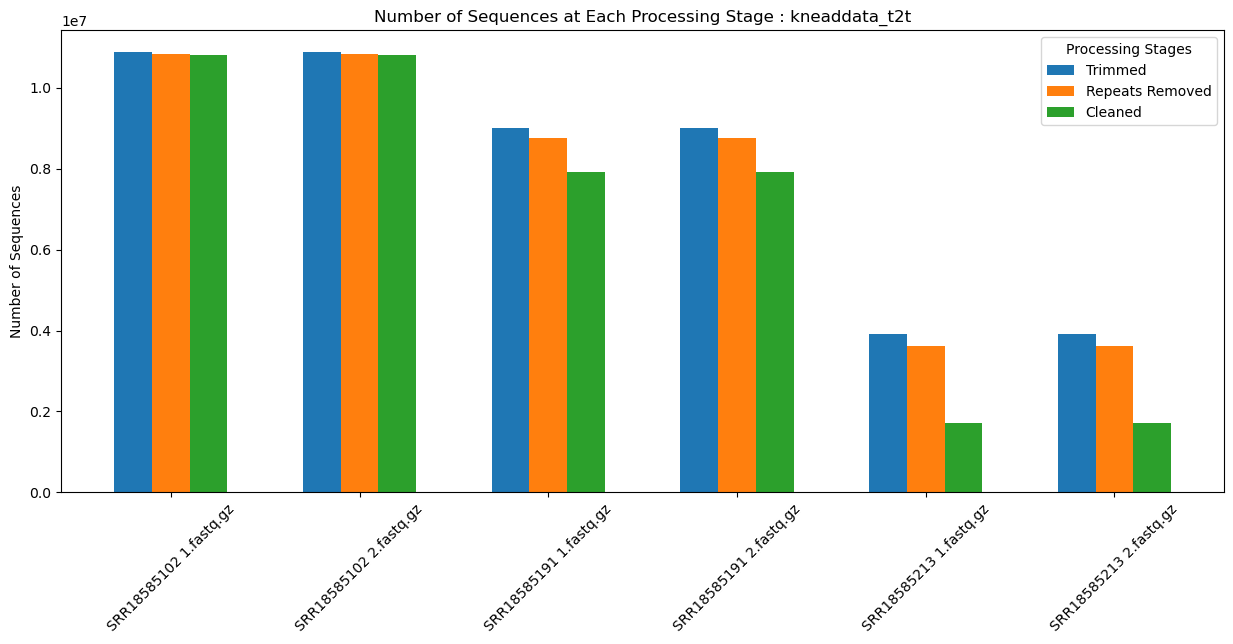
データによって処理が進むとガクンとリード数が低下しているものはありますが、ツールやホストゲノムによる違いは小さいように見えます(特定のファイルでリードが増えてる..)。
傾向として、予想に反してT2Tのリファレンスゲノムを使っているほうが、残存リードが多く、同じT2Tのリファレンスゲノムを使った場合では、hostileの方が残存リードが多いようです。
個人的にはマップできる領域が増えると、取りこぼしてたリードがマップされて、残存リードが減ると考えてました。
長くなってきたのでここらへんでひとまず終了。
収穫として、
- KneaddataでもT2Tリファレンスは使える
- Kneaddataだと最終的に残るホスト以外のリード数は、T2T > CRCh37
- ツール間だと最終的に残るホスト以外のリード数は、hostile > Kneaddata
ショートリードだとよほどの理由がない限りKneaddataで、ロングとショート両方とも同じツール使いたいなら、hostileがいいかなと思いました。
適当な比較記事でしたが、お付き合いいただきありがとうございました。
Infomation
- 自作PC
Discussion