PLINK2とPythonで集団遺伝学に入門する(1)データの前処理(HWE・アリル頻度分布)
はじめに
記事を書いた背景
筆者が業務上の都合で集団遺伝のデータを扱うことになったが、集団遺伝学の知識が皆無なので一から勉強したメモ。および、PLINK2を試したところ
- PLINKの資料は1についてがほとんどでPLINK2についての資料が乏しい
- PLINK1とPLINK2とではオプションの名称が大きく変化
- PLINK2は精力的に開発されておりしばしば破壊的更新が行われる結果、バージョン変更やエラーによりチュートリアルの内容が一部再現できない
- PLINK後の解析や図示にRが使われるが、筆者は宗教上の理由でRを使わない
のような問題に当たったので自分なりの解決方法を考えた。
つまり初心者が書いているのでやり方や説明が間違っている場合があります。
これを書いている合間にもバージョン変更が起きているが、本記事はPLINK2 Alpha6.2で確認している。
全体の章立て
複数の記事に分かれる。
- データの前処理(HWE・アリル頻度分布)(ここ)
- 連鎖不平衡と集団構造(仮)
- GWAS and eQTL(仮)
- 選択圧解析(仮)
参照
以下を読みながら記事を作成した。記事の流れはPLINK2の公式チュートリアルに沿っている。
- PLINK2の公式チュートリアル
https://www.cog-genomics.org/plink/2.0/tutorials/tutorial_setup - 遺伝統計学夏の学校講義資料(リンクは岡田随象研究室のHP;資料自体は毎年URLが変わるので・・・)
http://www.sg.med.osaka-u.ac.jp
Acknowledgement
二階堂雅人研究室の皆様。
PLINK2のファイル形式
PLINKは様々な集団遺伝解析を実行するモジュールをまとめた1つの大きなパッケージであり、集団遺伝解析では最も一般的に用いられているソフトウェアである。PLINKでは実行の効率化のために、独自形式のファイルを入力として取る。これは種のReferenceゲノムに対する個体の変異をコールしたデータ(多くはVCF形式)をPLINKに読み込ませることによって生成できる。
PLINK1の時代ではこのPLINK独自のファイルは.bim, .fam, .bed(bedtoolsのあれではない)というものであったが、PLINK2になってからフォーマットの大幅な刷新が行われ、.pgen, .pvar, .pfamという形式に変更された。もちろんPLINK2はPLINK1のデータ形式で実行することができ、またvcfファイル等PLINK形式のものでない形式でも直接入力にして解析することができる。しかしその場合は(たぶん)実行速度が劣っていたり、実行できる解析が限られる。
.pgen
個体ごとのgenotypeを保存したバイナリファイル。genotypeとは個体の持つ変異やフェーズの情報(複数のアリルが同じ親の染色体由来かどうか)など全般を指す。バイナリなので中身がどうなっているのかよくわからない。一応どのようなデータが格納されているかは--pgen-infoオプションで確認することができる。これにより
- Number of variants
- Number of samples
- Are all REF alleles 'known', 'provisional', or a mix?
- Maximum allele count for a single variant (exact value may require .pvar input)
- Are phased hardcalls present?
- Are dosages present? Are any of them explicitly phased?
が表示されるとのこと。
.pvar
各変異のメタ情報を以下のように格納している(ヘッダー略)。

これはバイナリではなくただのtsvなので弄ろうと思えば中身を弄ることができる。中身は#のヘッダー行に続いて変異の位置やID、Ref/Altなどの情報が保存されている。列の名前からわかるように、ヘッダーや列のほとんどはVCFの形式を引き継いでおり、VCFを簡略化したような形式である。最終ヘッダー行は列のラベルで必ず#CHROMから始まる。各列のラベルとしては以下のものを識別する。
- POS (base-pair coordinate)
- ID (variant ID; required)
- REF (reference allele)
- ALT (alternate alleles, comma-separated)
- QUAL (phred-scaled quality score for whether the locus is variable at all)
- FILTER ('PASS', '.', or semicolon-separated list of failing filter codes)
- INFO (semicolon-separated list of flags and key-value pairs, with types declared in header)
- FORMAT (terminates header line parsing)
- CM (centimorgan position)
.psam
個体ごとのメタ情報を以下のように格納している。

.pvar同様にバイナリではない。最終ヘッダー行は列のラベルで必ず#FID or #IIDから始まる。デフォルトのラベルとして以下のものが用いられる。
- IID (individual ID; required)
- SID (source ID, when there are multiple samples for the same individual)
- PAT (individual ID of father, '0' if unknown)
- MAT (individual ID of mother, '0' if unknown)
- SEX ('1' = male, '2' = female, 'NA'/'0' = unknown)
これ以外のラベルはすべてphenotypeのデータとして識別され、データ解析に用いることができる。例えば画像のSuperPopとPopulationの列は後から追加したものである。
PLINK2ファイルの生成
ヒトの遺伝的多様性を明らかにするプロジェクトである1000 Genomes Project(1KGP)で公開された、2500人あまり(現在はもっと多い)の全ゲノムシークエンスから作成されたgenotypeデータを用いて解析を行っていく。
1KGPのPLINK2形式のファイル(.pgen, .pvar, .psam)はすでにPLINK2の開発者たちが作ってくれているため、PLINK2 HPのResorucesのページから取得できる。
しかしこの記事ではより汎用的にPLINK2を用いることができるように、各PLINK2形式のファイルをマニュアルで生成する。
ヒト集団の遺伝情報の取得
すべてのヒト染色体のgenotypeデータを処理するのはチュートリアルとして重すぎてしまうので、ここでは一番データ容量が少ないchr22を用いる。1KGPが作成したchr22のvcfファイルは以下のftpサイトから入手できる。
wget https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/ALL.chr22.shapeit2_integrated_snvindels_v2a_27022019.GRCh38.phased.vcf.gz
VCFファイルへのrsidのアノテーション
遺伝的多様性研究をスムーズにするために、ヒトゲノム(Referenceゲノム)上で観察される変異にはその変異を一意に識別するためにrsidという識別子がつけられている。これはdbSNPというデータベースで管理され、世界で共通して用いられる。
ダウンロードしたvcfファイルは個体ごとのゲノムシークエンスデータから変異をコールした直後の状態であるため、このrsidがつけられておらず不便である。そこでdbSNPのアノテーションをダウンロードしてきてvcfファイルにrsidを追加する。vcfファイルにrsidを追加すればPLINK2で.pvarを生成するときにrsidが引き継がれる。
dbSNPのアノテーションはUCSC genome browserから取ってくる。


vcfファイルの各アリルにrsidを追加する。
awk -v OFS='\t' 'NR > 1{alt=substr($6, 1, length($6)-1);print $1,$2+1,$5,alt,".",$4}' dbSNP155.chr22.txt |\
perl -pe 's/chr//g' |bgzip -c > id_list.tsv.gz
tabix -s1 -b2 -e2 id_list.tsv.gz
bcftools annotate -a id_list.tsv.gz -c CHROM,POS,REF,ALT,-,ID \
-o chr22_annotated.vcf.gz ../ALL.chr22.shapeit2_integrated_snvindels_v2a_27022019.GRCh38.phased.vcf.gz
.pgen, .pvar, .psamの生成
rsidをつけたvcfファイルからPLINK2で.pgen, .pvar, .psamを生成する。
plink2 --vcf chr22_annotated.vcf.gz \
--make-pgen \
--snps-only \
--max-alleles 2 \
--set-missing-var-ids @:#_\$r_\$a \
--out hg38_chr22
- --make-pgen
.pgen, .pvar, .psamを生成する共通オプション。今回のようにvcfファイルなどから生成する場合のみならず、例えばフィルタリング後のファイルを生成するときにもこのオプションが共通して用いられる。 - --snps-only
一塩基多型以外の変異を除外する。今回は解析結果の説明が単純になるようにSNPのみを用いる。 - --max-alleles 2
PLINKの仕様上、1つの座位に複数の種類の変異が生じた場合(multiallelic)を扱えないため、multiallelicな変異を除外する。 - --set-missing-var-ids
rsidが割り振られていない変異もvcfファイル中には存在するため、そういった変異にユニークな名前を割り振る。今回は"chr":"locus"_"ref"_"alt"の規則性で名前をつける。 - --out
出力ファイル名を指定する共通オプションである。併用するコマンドによって出力ファイル名の拡張子は異なるため、詳しくは出力ファイル名のprefixを指定する共通オプションである。例えば--make-pgenを用いると.pgen, .pvar, .psamが生成されるため、--outはこれらのファイルのprefixを指定している。
これでvcfファイルからPLINK2形式のファイルhg38_chr22.pgen, hg38_chr22.pvar, hg38_chr22.psamが生成される。
しかし.psamには本来サンプルのメタ情報が入るはずだが、vcfファイルにはそういったメタ情報が含まれないため.psamの中身が空になる。そのため1KGPから.pedファイルおよびサンプルのメタ情報を取ってきてこれらをもとに.psamファイルを手動作成する。
.pedファイルのダウンロード
wget https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/integrated_call_samples_v3.20200731.ALL.ped
1KGPで用いられた個人のメタ情報は"Download the list"からダウンロードできる。ファイル名は"igsr_samples.tsv"。

.pedとigsr_samples.tsvをPythonで結合した後にpsamの手動生成する。
import pandas as pd
idx=pd.read_csv("hg38_chr22.psam",sep="\t",index_col=0)
ped=pd.read_csv("integrated_call_samples_v3.20200731.ALL.ped",sep="\t",index_col=1)
lis=pd.read_csv("igsr_samples.tsv",sep="\t",index_col=0)
data=pd.concat([ped,lis],axis=1,join="inner").loc[idx.index].reset_index()
out=data[["Family ID","#IID","Paternal ID","Maternal ID","Gender","Superpopulation code","Population code"]]
out.columns=["#FID","IID","PAT","MAT","SEX","SuperPop","Population"]
out.to_csv("hg38_chr22.psam",sep="\t",index=None)
これで生成したものが.psamの説明で見せた画像のものとなる。
Hardy-Weinberg平衡(HWE)とHWE検定
Hardy-Weinberg平衡
ある105人の集団で、ある遺伝子座にT/Cのアリルが観察され、個々人が持つ遺伝型TT/TC/CCの数が以下のような数値になったとする。
| genotype | TT | TC | CC |
|---|---|---|---|
| count | 66 | 25 | 14 |
| freq | 0.629 | 0.238 | 0.133 |
このときこの集団内に存在するTの数およびCの数はそれぞれT=2*66+1*25, C=2*14+1*25となるため、T/Cは以下のような頻度で集団中に存在することになる。
| allele | T | C |
|---|---|---|
| count | 157 | 53 |
| freq | 0.748 | 0.252 |
集団において新規の変異は常に生じているが、変異は交配によって集団内に確率的に広まり確率的に消滅する。このとき、その集団が均一な集団である場合、つまり雌雄間の交配に偏りがなくランダムと見なせて他の集団からの流出入もなく進化適応による遺伝情報の変化もないという理想状態の場合、ある遺伝子座のアリルA/aにおいてAの頻度をpとすると、遺伝型AA/Aa/aaの集団内での頻度はアリル頻度の単純な組み合わせである
に従う。これをHardy–Weinberg(ハーディー・ワインベルク/ハーディー・ワインバーグ)平衡と言う。
Hardy–Weinberg平衡検定(HWE検定)
実際、集団内に存在する多くのアリルはこのHardy–Weinberg平衡にある(はずである)。Hardy–Weinberg平衡から逸脱する変異は、変異のコールエラーまたは何かしらの選択圧がかかっている変異であることが考えられる。多くの統計解析手法は集団が均一であることを前提とするため、こうした変異はクオリティーコントロールの一環として除外される。
各アリルがHardy–Weinberg平衡から逸脱しているかの検定を、そのままの名前でHardy–Weinberg平衡検定(HWE検定)という。HWE検定の方法は単純で、まず遺伝型の実測値からアリルの頻度を計算し、アリルの頻度から各遺伝型の頻度の理論値を算出する。そしてこの理論値が実測値とどれくらい乖離しているのかを
例えば上記で用いたT/Cのアリルの場合、それぞれの頻度を0.748、0.252と算出した。Tの頻度をp=0.748と置くと、遺伝型TT/TC/CCの頻度の理論値はそれぞれ以下のように算出できる。
| genotype | TT | TC | CC |
|---|---|---|---|
| 数式 | |||
| 頻度の理論値 | 0.560 | 0.377 | 0.064 |
この頻度の理論値に集団サイズ(=105)をかけると各遺伝型を持つ個体数の期待値となる。次にこの理論値と実測値が有意に逸脱しているかを検定するが、Fisher's exact testを行うと激しい階乗の計算になるためここでは
| genotype | TT | TC | CC |
|---|---|---|---|
| 期待値 | 58.8 | 39.585 | 6.72 |
| 実測値 | 66 | 25 | 14 |
変数はpのみなので自由度1の
PLINK2での実行
PLINK2でのHWE検定は--hardyオプション1つで実行してくれる。
plink2 --pfile hg38_chr22 --hardy --out chr22_HW
- --pfile
解析に用いるPLINK2ファイルのprefixを指定する共通オプション。指定したprefixに続く.pgen,.pvar,.psamファイルを随時読み込んでくれる。 - --hardy
HWE検定をやります。
結果はchr22_HW.hardyというファイルに出力される。この集団(このデータでは世界中の2500人あまり)における各アリルや遺伝型の頻度、それらがHWEに対してどれくらいのp-valueで乖離しているのかが各列に記されている。

- CHROM
染色体(or scaffold) - ID
.pvarに表記されたSNPのID - A1
Reference genomeの配列 - AX
変異 - HOM_A1_CT
A1|A1の数 - HET_A1_CT
A1|AXの数 - TWO_AX_CT
AX|AXの数 - O(HET_A1)
A1|AXの頻度 - E(HET_A1)
A1とAXの頻度から計算したA1|AXの頻度の期待値 - P
p-value。たぶんFisher's exactで計算している。adjusted p-valueを使うべきな気がするが、これは補正後の値なのかどうかはわからない。たぶん補正前の値。
ここで各アリルの遺伝型がどのような分布になっているのかを図示してみる。
Hardy-Weinberg平衡状態のとき、アリルA/aの片方の頻度をpとしたとき、ヘテロな遺伝型(Aa)の頻度は2p(1-p)となる。そのためx軸にp, y軸にヘテロな遺伝型の実測値をプロットするとどれくらいのアリルがHardy-Weinberg平衡に達しているのかを可視化できる。
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
hwe=pd.read_csv('./chr22_HW.hardy',sep='\t')
pop_size=hwe[['HOM_A1_CT','HET_A1_CT','TWO_AX_CT']].sum(axis=1)*2
#alternative allele頻度
hwe['afreq']=(hwe['HET_A1_CT']+2*hwe['TWO_AX_CT'])/pop_size
x=hwe['afreq']
y=hwe['O(HET_A1)']
plt.scatter(x,y,s=1,alpha=0.1)
plt.xlabel('alt_freq')
plt.ylabel('observed_het_freq')
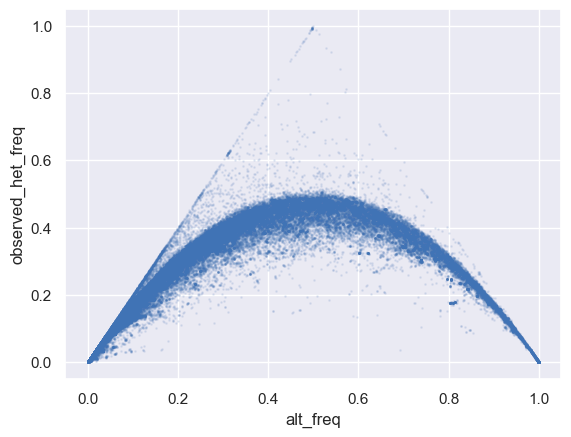
この図にHerdy-Weinberg平衡時の理論値(つまりy=2p(1-p)の二次関数)を載せてみる。
p=np.linspace(0,1,50)
plt.plot(p,2*p*(1-p),linestyle='dashed',c='black',linewidth=1)

もしほとんどのアリルがHerdy-Weinberg平衡にあるのならば、アリルの分布は理論値y=2p(1-p)の二次関数に対して対称になるはずである。しかし、実際にはほとんどのアリルのヘテロの頻度は理論値の曲線から逸脱しており、特に理論値を下回っているのがわかる。つまり各アリルはホモで存在することに強くバイアスがかかっているということだが、これは遺伝型がランダムに配分される状況ではありえない。これはこの集団がHerdy-Weinberg平衡に従っていないことを意味する。
当然のことだが、この状態で行ったHWE検定のp値でフィルタリングすると多くのアリルが誤って捨てられてしまう。
mask=hwe['P']>0.05
#qualified
plt.scatter(x[mask],y[mask],s=1,c='yellow',alpha=0.1)
#not qualified
plt.scatter(x[~mask],y[~mask],s=1,alpha=0.1)

今回用いたデータは世界中から集めたヒトのデータだが、世界中の人々はもちろん完全ランダムにパートナーを選んでいるわけではなくそれぞれ地理的に近い地域内で交配する。そのためこのデータはHerdy-Weinberg平衡が要求する「均一な集団」ではない。アリルがホモで存在することに強くバイアスがかかっているように見える理由としては、「世界」というのは複数のヒト集団を含んでいるため、ある集団に存在するアリルがHerdy-Weinberg平衡に達していても、その他の集団ではそのアリル自体が存在しないため、世界で合計するとホモの遺伝型が多くカウントされてしまうと考えられる。集団遺伝解析はこのようなグローバルな集団ではなく、適切なローカルな集団ごとに行われなければならない。
このように、解析をする前にヘテロな遺伝型の頻度がどれくらいHWEの理論値に近いかを比較することによって、自分が扱おうとしているデータが適切な集団であるかを調べることができる。これもクオリティーチェックの一部と言えるが、フィルタリング云々よりもおそらく大事な前処理である。
また、これらヘテロ頻度が過小評価されているアリル群以外にもいくつかの点が明らかにHWEを逸脱しているように見える。例えば図中で三角形の輪郭上に薄っすらとある点は、アリル頻度とヘテロな頻度が比例定数2で比例していることになるため、アリルの片方がすべてヘテロで存在しホモでは存在しないアリルということになる。確率的にこれはあり得ず、これらのアリルはコールエラーによるものである。例えば異なる遺伝子座にある反復配列が同じ遺伝子座に誤ってマッピングされてしまいアリルとしてコールされてしまったことなどが考えられる。
Subpopulationでの実行
グローバルな集団のデータを用いてもHWE検定がうまく機能しないことを前章で可視化して確認した。そこで今度はローカルな集団に区切って実行する。1KGPのデータは個体の出身やどの祖先集団に属するかのアノテーションを持っており、この記事ではそれを以下のように.psamファイルに出力している。

このPopulationの列のうち、JPT(Japan Tokyo)出身のヒトに限定してHWE検定を実行してみる。
plink2 --pfile hg38_chr22 --hardy --out chr22_HW_JPT --keep-if Population==JPT
- --keep-if
指定した条件にマッチするサンプルに対して実行するオプション。逆は--remove-if
同様にアリル頻度とヘテロな遺伝型の頻度をプロットする。
hwe_jp=pd.read_csv('/Users/z_zhang/gdrive/InoueLab/1KGP/chr22_HW_JPT.hardy',sep='\t')
size=hwe_jp[['HOM_A1_CT','HET_A1_CT','TWO_AX_CT']].sum(axis=1)*2
hwe_jp['afreq']=(hwe_jp['HET_A1_CT']+2*hwe_jp['TWO_AX_CT'])/size
x=hwe_jp['afreq']
y=hwe_jp['O(HET_A1)']
plt.scatter(x,y,s=1)
plt.xlabel('alt_freq')
plt.ylabel('observed_het_freq')
p=np.linspace(0,1,50)
plt.plot(p,2*p*(1-p),linestyle='dashed',c='black',linewidth=1)

JPTはグローバルな集団よりも均一な集団であるため、グローバルな集団で見られていたヘテロな遺伝型の頻度に働いていた下方向へのバイアスがなくなり、各アリルは二次関数y=2p(1-p)に対して対称に分布することがわかる。
もちろんこの状態でp値のフィルタリングを行えばちゃんとフィルタリングできる。
mask=hwe_jp['P']>0.05
#qualified
plt.scatter(x[mask],y[mask],s=1,c='yellow',alpha=0.1)
#not qualified
plt.scatter(x[~mask],y[~mask],s=1,alpha=0.1)

逆に言えば、このようにsubpopulationがわかっている状況でないと集団解析がうまくできないことになる。事前に集団構造がわからない場合は、次回の記事で行う集団構造の可視化によってデータがどのような集団構造を持つのかを予め検知する。
アリル頻度の分布
PLINK2でのアリル頻度の計算と可視化
GWASなどにおけるデータの前処理として、アリル頻度自体でフィルタリングするのも一般的である。これはアリル頻度のごくごく小さいもの(<1-10%程度)はSNPコールのエラーである可能性が高いことや集団に定着しない変異と考えられるため解析にはほとんど影響を与えないことなどから解析から省いてしまうというものである。
PLINK2でのアリル頻度の計算は簡単で、--freqというオプション1つで行う。ここではHWE検定のときと同様にまずグローバルな集団(ALLとラベルする)でアリル頻度を計算し、ローカルな集団でカウントしたときとの違いを比べる。
plink2 --pfile hg38_chr22 --freq --out chr22_ALL_freq
結果は.afreqというファイルに出力される。

- ALT_FREQS
Refの配列に対する変異(alternative)の頻度。つまりはアリル頻度なのだが、alternativeがマイナーとは限らないのでalternative allele freqと記述される。
アリル頻度を0.01%刻みのヒストグラムとして可視化してみる。
afreq=pd.read_csv('./chr22_freq.afreq',sep='\t')
bin_width = 0.01
bins = np.arange(0.01, 1 + bin_width, bin_width)
plt.hist(afreq['ALT_FREQS'],bins=bins)
plt.yscale('log')

これを見るとfreqが0および1付近にピークがあることがわかる。アリル頻度でフィルタリングするときにはこのようなピークを省く。
今度はいくつかのsubpopulationにおけるアリル頻度を計算してみる。これはHWE検定のときと同様に--keep-ifでできる。
subpops=("LWK" "ASW" "CHB" "JPT")
for subpop in "${subpops[@]}"
do
plink2 --pfile hg38_chr22 --freq --out chr22_${subpop}_freq --keep-if 'Population=='$subpop''
done
- LWK
Luhya in Webuye, Kenya - ASW
African Ancestry in Southwest US - CHB
Han Chinese in Beijing, China
それぞれの集団で各アリル頻度がどのように変化しているのかをpairwiseの散布図で可視化する。
g=sns.pairplot(afreq_div, kind="scatter", plot_kws={'alpha': 0.1, 's': 1}, corner=True, diag_kind=None)
for i in range(len(afreq_div.columns)):
g.axes[i, i].remove()

これを見るとグローバルな集団ALLでのアリル頻度と各subpopulationの頻度はある程度相関するものの、相関しないものもある。そのため、例えばALLの頻度10%以下のアリルを除いた場合、あるsubpopulationでは頻度が50%のアリルを排除してしまうかもしれない。そのため、HWE検定のときと同様にアリル頻度のフィルタリングもsubpopulationを考慮しなければならない。
また、CHB vs. JPTを見ると散布図はほとんど直線であることから、地理的に近い集団はやはり遺伝的にも近いことがわかる。
HWE検定 vs. アリル頻度
今度はHWE検定の結果とアリル頻度の関係を可視化して見よう。JPTでHWE検定を行ったデータがあるので、これをそのまま利用してアリル頻度vs.HWE検定のp値で散布図を描く。
plt.scatter(hwe_jp['afreq'],-np.log10(hwe_jp['P']),s=1,alpha=0.1)
plt.xlabel('JPT_allele_freq')
plt.ylabel('-log10(HWE p-value')

簡単に言えば今回の図は以前の図(HWE検定のアリル頻度vsヘテロ頻度の図)が二次関数に曲がっていたのをまっすぐにした感じになる。HWE検定の結果でフィルタリングすることを考えたとき、アリル頻度がごく僅かなアリルでもHWE検定が有意でないSNPが存在する。また、アリル頻度でフィルタリングすることを考えたとき、HWE検定の結果が有意なSNP(例えばp<0.05等)はアリル頻度が高いものでも見られるため、アリル頻度のみでのフィルタリングではこれらを排除することはできない。そのため、HWE検定とアリル頻度の両方でのフィルタリングが必要がある。
結論
以上により、集団データをフィルタリングするときにはHWE検定とアリル頻度の両方に加え、集団構造を考慮しなければならないことを可視化した。最終的なフィルタリングコマンドとして、例えばHWE検定のp値(>0.05:--hwe)とアリル頻度(>5%:--maf)でJPTのsubpopulationが持つアリルをフィルタリングする場合は次のようになる。
plink2 --pfile hg38_chr22 --make-pgen --hwe 0.05 --maf 0.05 --keep-if Population==JPT --out chr22_JPT_qcd
これによりクオリティーチェックにクリアしたアリルとJPTのサンプルのみを含む.pgen, .pvar,.psamが生成される。
補足説明:Ancestry group(≠人種)について
今回はわかりやすさのためにローカルな集団としてJPTを用いたが、実際はJPTほど細かくする必要はなく、1KGPの定めるSuper population(Ancestry group:祖先グループ)単位で解析をすれば十分である。これは日本語の「人種」のニュアンスに近いヒト集団の区分である。しかし以前定められたモンゴロイドやコーカソイド等の人種区分は身体的特徴によって分けられている一方、ゲノムから得られた情報を解析すると必ずしもこの身体的特徴によってヒトは区分されないことが示されている。そのため、代わりに新しくゲノムと地理的情報に基づいた区分として現在ではAncestry groupという概念が用いられる。
1KGPの定めるSuper populationとしては現在AFR(アフリカ),AMR(中南米),EAS(東アジア),EUR(ヨーロッパ),SAS(南アジア)の5つが用いられる。JPT等のより細かいpopulationはこのどれか1つのSuperPopに属するとは限らず、いくつかのAncestry groupの混合として表現される場合もある。
例えばSuperPopのEURについて先程のalt頻度vs.ヘテロ頻度をプロットすると理論値y=2p(1-p)とほぼ線対称になる。しかしよく見ると点が少しだけy=2p(1-p)の下に偏っていることから、SuperPopも複数のローカルな集団の集合であることを示唆している。ただ、p>0.05で切る限りは大丈夫そう。


Discussion