【R】遺伝子発現をggplot2でシンプルな棒グラフ作成 - ①
はじめに
目標
Real-Time qPCRのcsvデータをRstudioに読み込ませて、棒グラフの描画をしてもらいます。よく論文で見るような、シンプルな見た目かつ、エラーバーや有意差検定の結果まで一気に表示してくれるfigureの作成を最終目標にしていきます。
今回はワンステップおいて、エラーバーのみのシンプルな棒グラフの描画を目標にします。
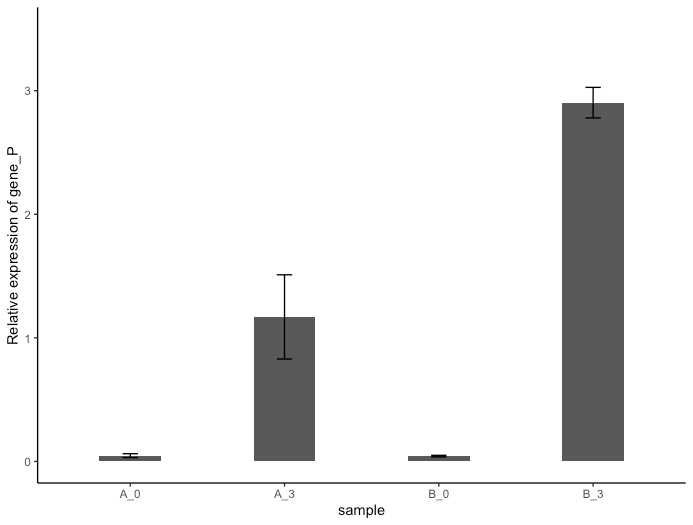
今回の記事では、こんな感じのグラフを描画できるようになります。
(本当はグラフタイトル、ストレス処理の有無、有意差検定とかつけてみたい)
以前はExcelで棒グラフの作成をしていましたが、Rの勉強モチベーションが高かったので、今のうちに冷めやすい鉄を打っておきました。
実験条件と用いるデータ
今回のfigは、植物が特定のストレスを受けた際に遺伝子発現をどのように変化させているのか、棒グラフで示します。
本実験は、モデル植物(シロイヌナズナ)のストレス応答について調べます。
植物は動物と違い、環境ストレス(乾燥、温度、塩、光など)に晒されても、ストレスのない快適な場所へ移動することができません。そのため植物はストレスを感知して、植物体内の生理変化(植物ホルモンや遺伝子発現の調節)を引き起こしてその場で耐えます。
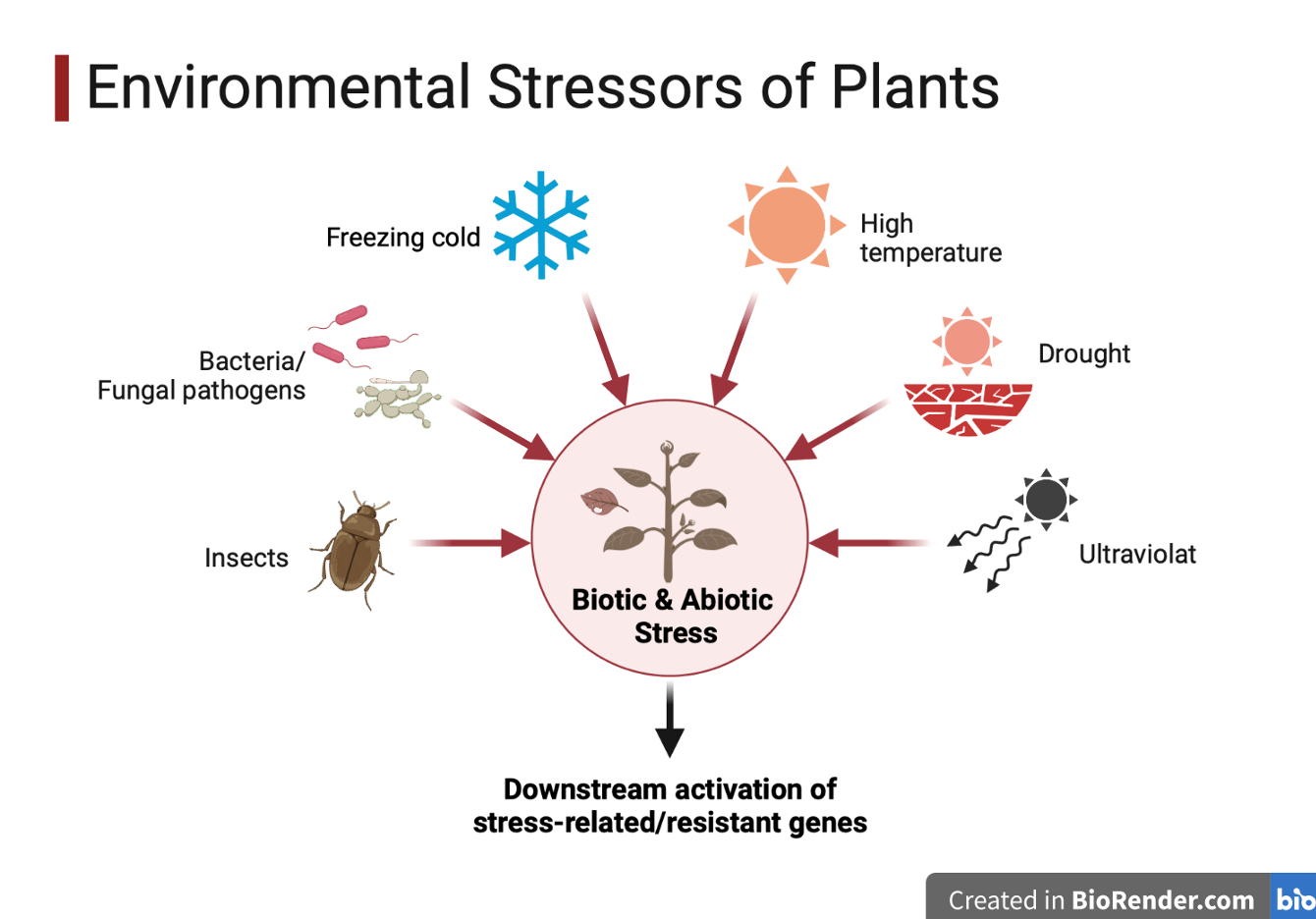
BioRenderさんで作成
その遺伝子発現量がストレス処理前後でどう変化しているのかを調べるため、特定のストレス処理を行った植物体からRNAを抽出、cDNA合成、そしてストレス誘導性遺伝子をマーカーとしてRT-qPCRを行いました。
以下、今回のグラフ作成に使っていくデータになります。ご活用ください。
sample_treatment-time,gene
A_0,0.043122871
A_0,0.063175676
A_0,0.032600745
B_0,0.050309051
B_0,0.040279368
B_0,0.038257683
A_3,0.992119453
A_3,0.953639314
A_3,1.562149158
B_3,2.883036936
B_3,2.791183295
B_3,3.036106124
データの見方
sample_treatment-timeのcolumn(列)は、アンダーバーを挟んで左側がサンプル名(エコタイプ名とか)、右側にストレス処理時間です。
また、geneのcolumnは、ストレス応答性遺伝子(gene_Pとしましょう)の発現量です。厳密に言うと、ハウスキーピング遺伝子(ACT2)の発現量でNormalizeしたrelative expressioionですが。
一番下のrow(行)を例に見てみると…
B_3,3.036106124
これは、エコタイプBの植物にストレスを3日間処理した結果、gene_Pの発現量が、ACT2に比べて約3倍だったということです。
要求package
- ggplot2
- dplyr
動作環境
- MacBook Air(M1, 2020), macOS Monterey 12.0.1
- RStudio, "Ghost Orchid" Release for macOS Mozilla/5.0 (Macintosh; Intel Mac OS X 12_0_1)
1. ワーキングディレクトリの設定とパッケージのロード
忘れないうちに、初めにやっておきます。
setwd('~/Desktop/Zenn/bar-graph')
#グラフ化したいデータのcsvファイルが入っているディレクトリをワーキングディレクトリに設定
僕はDesktopにおいてある、Zennフォルダ下のbar-graphフォルダに、sample-gene.csvを置いていたので、そこにsetwd()で仕事場を指定。
続いて、パッケージのロードのためにlibrary()を実行します。ロードするパッケージは以下2つ。
- ggplot2
- dplyr
未インストールの方はinstall.package()であらかじめインストールしておきましょう。(インストールは初回のみ)
library(ggplot2) #グラフの描画に使う
library(dplyr) #データの加工に使う
2. csvファイルの読み込みと加工
次に実験データの入っているcsvファイルをRStudioに読み込ませます。
d <- read.csv("sample-gene.csv", header = T)
上の操作では、read.csv()を用いて読み込んだcsvファイルを、dという文字に代入する操作をしています。(dはなんの文字を指定しても良いです。僕の場合は、短い方が楽なのでdataの頭文字を指定しています。)
header = Tはデータの1行目がサンプル名とか、数値データじゃない時に指定してあげます。1行目から数値データの時にはheader = Fを指定します。
dを入力、実行してあげると、Consoleに表が出力されます。(実行結果↓)

続いて、データフレームを作成します。今のままだと少し列名が長いので、シンプルにしてコード量を減らします。あと、geneだと遺伝子名とかと混同してしまいそうなので、発現量(数値データ)だとわかるようにexpressioionに変えてみました。
d1 <- data.frame(sample = d$sample_treatment.time, expression = d$gene)
d1を入力、実行してあげます。(実行結果↓)

しっかり反映されています!
3. 平均値・標準偏差の算出
dplyrのgroup_by関数とsummarise_all関数
続いてグラフとエラーバーの描画のために、sampleごとに平均値と標準偏差を算出しておきます。
ここで、dplyrの機能、group_by()関数と、summarise_all()関数が役に立ちます。
d_mean_sd <- d1 %>% #1行目
group_by(sample) %>% #2行目
summarise_all(list(mean = mean, sd = sd)) #3行目
上のコードの意味は、
-
d1のデータをgroup_by()関数でsampleごとにまとめて(2行目) -
summarise_all()でサンプルごとの平均値(mean)と標準偏差(standard deviation)を算出(3行目)
これらの操作を、d_mean_sdに格納しています。(1行目)
試しにd_mean_sdを実行してみると…(実行結果↓)

サンプルごとに、平均と標準偏差が求まったテーブルが出来上がりました!
ついでに、列の名前もわかりやすいものに変更しておきます。
colnames(d_mean_sd) <- c("sample", "mean.expression", "sd.expression")
エラーバーの準備
さらに、グラフにエラーバーを描画させるため、aes()関数で、エラーバーに“長さ”という属性を付して(僕のaes関数への理解では、こういう意味だと思っています。もしかしたら、もしかしなくても間違っているかもしれないです。)、さらにこれをerrorという函に格納しておきます。
errors <- aes(ymin = mean.expression - sd.expression, ymax = mean.expression + sd.expression)
ここまでで、グラフ描画のお膳立てが終わりました〜!
4. グラフ作成
早速、ggplotの機能を使っていきます。
g <- ggplot(d_mean_sd, aes(x = sample, y = mean.expression)) +
geom_bar(stat = "identity")
plot(g)
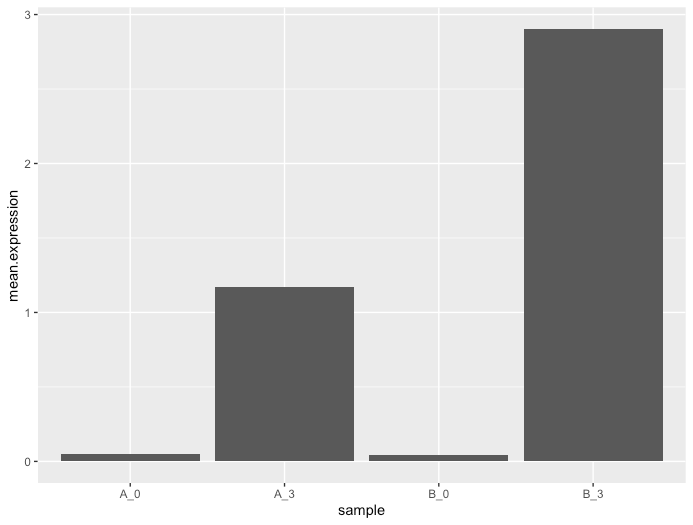
実行結果↑
ggplot()の後ろに + で色々付け足すことで、グラフの見た目を変えることができます(追加パッケージとかもあるみたい)。例えば、+geom_errorbar()でグラフにエラーバーを付けることができます。
g <- ggplot(d_mean_sd, aes(x = sample, y = mean.expression)) +
geom_bar(stat = "identity") +
geom_errorbar(errors)
plot(g)
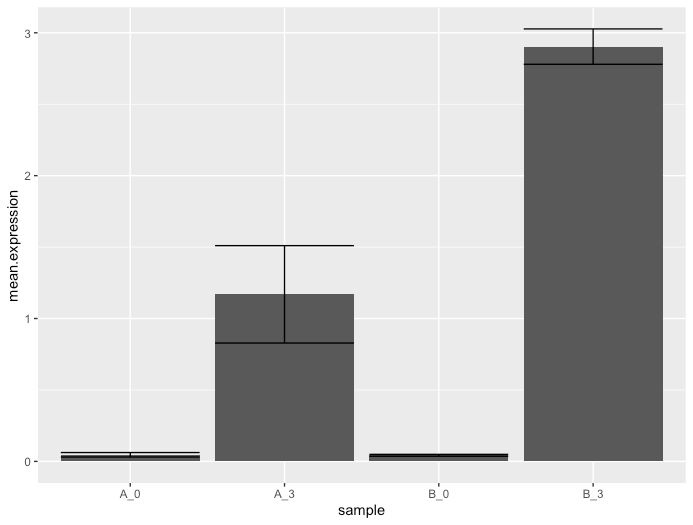
とりあえず描画はできましたが、謎の灰色みがかった背景に、太い棒とエラーバーがとにかくダサいです。もう少しだけ手を加えます。
g <- ggplot(d_mean_sd, aes(x = sample, y = mean.expression)) +
geom_bar(stat = "identity", width = 0.4) +
geom_errorbar(errors, width = 0.1) +
scale_y_continuous(name = "Relative expression of gene_P") +
theme(axis.title.x = element_text(size=10, family = "Arial"),
axis.title.y = element_text(size=10, family = "Arial"),
axis.text.x = element_text(size=10, colour = 1, family = "Arial"),
axis.text.y = element_text(size =10, colour = 1, family = "Arial")) +
theme_classic()
plot(g)
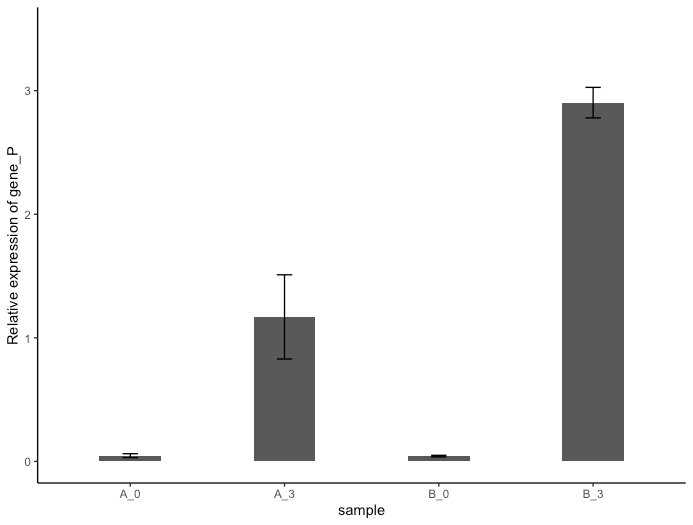
体裁は好みがあると思うので、色々とggplotの中身をいじってTPOに合わせてグラフ描画しましょう。
おわりに
今回は、Rのggplot2を使ってシンプルなグラフの描画をしてみました!
有意差検定の結果も同時に出力できるコードを書けたら、また記事を書くかもしれません。
実験結果
一応、実験条件についてお話したので、今回の個人的な出力データの解釈についても書いておきます。
(今回の解析では、検定試験を行っていませんが)ecotype AもBも、ストレス無処理のサンプルに比べて、ストレス処理3日後のサンプルの方でgene Pの発現レベルは増加している傾向が見られました。
gene Pは、シロイヌナズナのある特定のストレス応答のマーカー遺伝子として用いられる、ストレス応答シグナリングの下流因子で、この遺伝子の発現上昇はストレスに耐えようとしたかどうかの結果と解釈できます。(遺伝子発現は秒単位で起こるのに、ストレス処理後3日間もおいているのはgene Pがほぼ最下流だから)
つまり、ecotype A, Bともにストレス応答をしていますが、その程度はBの方がより高い(要検定)(気がします)
今回の全R script
setwd('~/Desktop/Zenn/bar-graph')
library(ggplot2)
library(dplyr)
d <- read.csv("sample-gene.csv", header = T)
d1 <- data.frame(sample = d$sample_treatment.time, expression = d$gene)
d_mean_sd <- d1 %>%
group_by(sample) %>%
summarise_all(list(mean = mean, sd = sd))
colnames(d_mean_sd) <- c("sample", "mean.expression", "sd.expression")
errors <- aes(ymin = mean.expression - sd.expression, ymax = mean.expression + sd.expression)
g <- ggplot(d_mean_sd, aes(x = sample, y = mean.expression)) +
geom_bar(stat = "identity", width = 0.4) +
geom_errorbar(errors, width = 0.1) +
scale_y_continuous(name = "Relative expression of gene_P") +
theme(axis.title.x = element_text(size=10, family = "Arial"),
axis.title.y = element_text(size=10, family = "Arial"),
axis.text.x = element_text(size=10, colour = 1, family = "Arial"),
axis.text.y = element_text(size =10, colour = 1, family = "Arial")) +
theme_classic()
plot(g)
Discussion