【R】遺伝子発現をggplot2で論文体裁な棒グラフ作成 - ②
はじめに
前回の記事の続き的な記事になると思います。
今回のR script作成にあたって、Aesthetic errorsなるものに相当苦しめられました。これを回避するために、何度もdataframeを定義し直すことになってしまって、少しコードが汚いです。
どなたか上手いやり方を教えてください。
スクリプトを書き直したので少しは可読性が高まったと思います。(2022/12/2)
目標
記事のコンセプトとしては、論文体裁な遺伝子発現量のグラフを、Rだけで作成できるようになることです。
より具体的には、遺伝子発現(Real-Time qPCR)のcsvデータをRStudioに読み込ませて、よく論文で見るようなシンプルな見た目かつ、エラーバーや有意差検定(多重比較)の結果まで載っているfigureの作成ができるコードを書いていきます。

今回の記事では、こんな感じのグラフを描画できるようになります。
前回のものと比べると、論文図表感がめちゃくちゃ増してませんか??
実験条件
(前回の記事と全く同じなので、読んだ方は読み飛ばしてください。)
#使用する実験データ ←ページ内リンク
今回のfigは、植物が特定のストレスを受けた際に遺伝子発現をどのように変化させているのか、棒グラフで示します。
本実験は、モデル植物(シロイヌナズナ)のストレス応答について調べます。
植物は動物と違い、環境ストレス(乾燥、温度、塩、光など)に晒されても、ストレスのない快適な場所へ移動することができません。そのため植物はストレスを感知して、植物体内の生理変化(植物ホルモンや遺伝子発現の調節)を引き起こしてその場で耐えます。
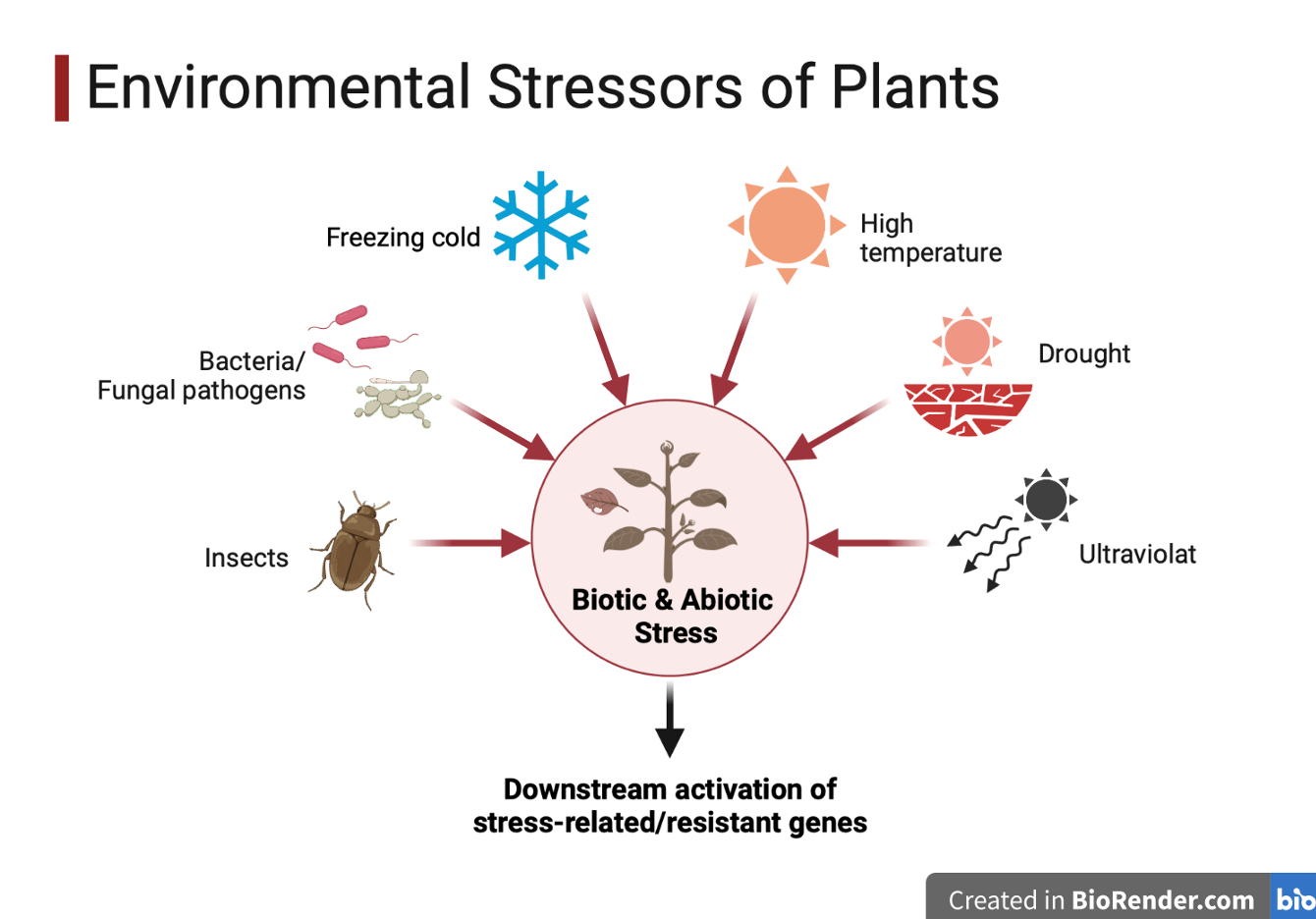
BioRenderさんで作成
その遺伝子発現量がストレス処理前後でどう変化しているのかを調べるため、特定のストレス処理を行った植物体からRNAを抽出、cDNA合成、そしてストレス誘導性遺伝子をマーカーとしてRT-qPCRを行いました。
使用する実験データ
以下、今回のグラフ作成に使用するcsvデータになります。
前回のものと少し違うので注意してください!
sample,time,gene
A,0 day,0.043122871
A,0 day,0.063175676
A,0 day,0.032600745
A,3 days,0.992119453
A,3 days,0.953639314
A,3 days,1.562149158
B,0 day,0.050309051
B,0 day,0.040279368
B,0 day,0.038257683
B,3 days,2.883036936
B,3 days,2.791183295
B,3 days,3.036106124
データの見方
これも前回の記事とほぼ同じですが、念の為に書いておきます。

sample-time-geneのcolumn(列)は、ハイフン(-)を挟んで左側がサンプル名(ecotype名)、真ん中がストレスの処理時間、右側がストレス応答性遺伝子(gene_Pとしましょう)の発現量です。厳密に言うと、ハウスキーピング遺伝子(ACT2)の発現量でNormalizeしたrelative expressioionです。
一番下のrow(行)を例に見てみると…
B,3,3.036106124
これは、エコタイプBの植物にストレスを3日間処理した結果、gene_Pの発現量が、ACT2に比べて約3倍だったということです。
要求package
- ggplot2
- dplyr
- multcomp
動作環境
以下の環境で動作確認済みです。
- MacBook Air(M1, 2020), macOS Monterey 12.0.1
- RStudio, "Ghost Orchid" Release for macOS Mozilla/5.0 (Macintosh; Intel Mac OS X 12_0_1)
1. ワーキングディレクトリの設定とパッケージのロード
例によって、忘れないうちにやっておきます。
setwd("~/Desktop/Zenn/bar-graph_2")
#グラフ化したいデータのcsvファイルが入っているディレクトリをワーキングディレクトリに設定
僕はDesktopにおいてある、Zennフォルダ下のbar-graph_2フォルダに、sample-time-gene.csvを置いていたので、そこにsetwd()で仕事場を指定。
続いて、パッケージのロードのためにlibrary()を実行します。今回ロードするパッケージは以下3つ。
- ggplot2
- dplyr
- multcomp
未インストールの方はinstall.package()であらかじめインストールしておきましょう。(インストールは初回のみ)
library(ggplot2) #グラフの描画に使う
library(dplyr) #データの加工に使う
library(multcomp) #検定に使う
2. csvファイルの読み込みと加工
次に実験データの入っているcsvファイルをRStudioに読み込ませます。
d <- read.csv("sample-time-gene.csv", header = T)
上の操作では、read.csv()を用いて読み込んだcsvファイルを、dという文字に代入する操作をしています。(dはなんの文字を指定しても良いです。僕の場合は、短い方が楽なのでdataの頭文字を指定しています。)
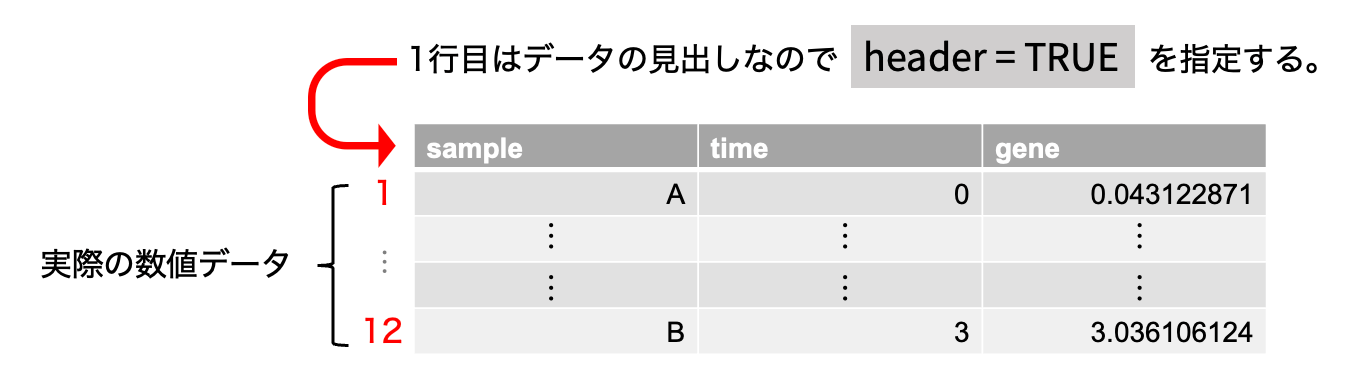
header = Tはデータの1行目がサンプル名とか、数値データじゃない時に指定してあげます。1行目から数値データの時にはheader = Fを指定します。
3. 検定用のデータフレーム作成〜Tukeyの検定
まず、グラフに描画する検定結果を先に計算します。
グラフの描画を、ストレス無処理/処理群で色分けしたいのでわざわざsample(ecotype名)とtime(ストレスの処理時間)でcolumnを分けてデータを作っています。しかし実のところは、統計解析をするにはまとまっていた方が都合が良いです。
というわけで、dplyrのmutate()関数をつかってsampleとtimeをくっつけた新しいcolumnを作ります。
検定用データフレーム作成
d1 <- mutate(d, sample_time = paste(d$sample, d$time)) #1行目
d_tukey <- data.frame(sample_time = d1$sample_time, ex = d1$gene) #2行目
d_tukey$sample_time <- factor(d_tukey$sample_time) #3行目
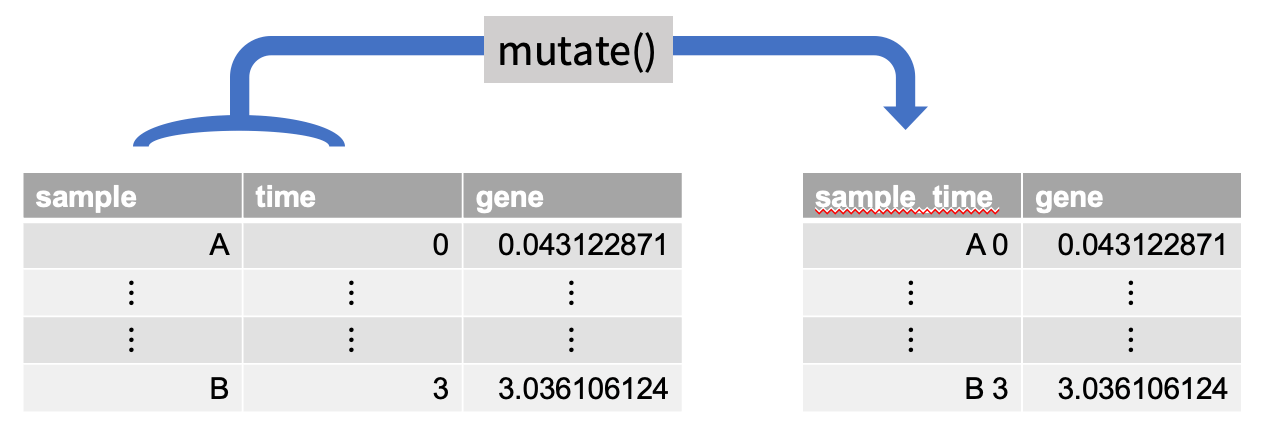
1行目の
mutate()関数のイメージ
2行目: d_tukeyに、新しく作成したsample_timeと、遺伝子発現の定量値をexとして格納しました。
3行目: 後に使用する関数がsample_timeを正しく認識できるようにデータの持つ属性を編集しています。
d_tukeyを実行して、データを確認してみます。↓
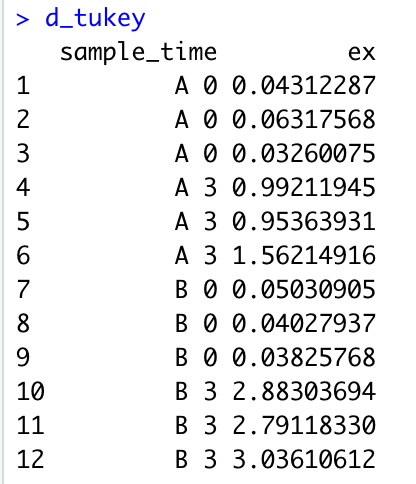
ここまでで、念のためにTukey検定ができるかも確認しておきましょう。
TukeyHSD(aov(ex ~ sample_time, data = d_tukey))

ちゃんとできていました〜
Tukeyの検定
あとは、【R】multcompで多重比較結果をアルファベット表示させるでやったように、検定結果をアルファベットで出力します。
res <- aov(ex ~ sample_time, data = d_tukey)
tuk <- glht(res, linfct = mcp(sample_time = "Tukey"))
mltv <- cld(tuk, decreasing = F)
abc <- mltv[["mcletters"]][["Letters"]]
以前の記事では書いていませんでしたが、実はこの検定結果(mltv)は、["mcletters"]][["Letters"]という場所にアドレスされています。
なので、abc <- mltv[["mcletters"]][["Letters"]]これを実行してあげることで、検定の結果のアルファベットを、abcという函に格納することができます!(これをグラフ描画に使います。)
4. グラフ描画準備
続いて、グラフ描画用のデータフレームを作っていきます。
僕たちが作りたいグラフの作成に必要な情報(Matrixで用意したいもの)は、
- sample名(ecotype AとかBとか)
- ストレス無処理/処理の条件(0 dayとか3 daysとか)
- 遺伝子発現の平均値
- 標準偏差(エラーバーつけるため)
- 有意差を示すアルファベット
dplyrのgroup_by関数とsummarise_all関数
まずはグラフとエラーバーの描画のために、遺伝子発現の定量値について、平均値と標準偏差を算出しておきます。
dplyrの機能、group_by()関数と、summarise_all()関数を使いましょう。
d_stat <- d %>%
group_by(sample, time) %>%
summarise_all(list(mean = mean,
sd = sd))
上のコードでは、d1のデータをgroup_by()関数でsample、timeごとにまとめてsummarise_all()関数に渡し、サンプルごとの平均値(mean)と標準偏差(standard deviation)を算出したものをd_statに格納しています。

d_statの実行結果
こんな感じのテーブルになってます。
グラフ描画のためのデータフレーム作成
d_statをもう少し加工して、グラフ描画に使える一つのデータフレームを作ります。
(美しくないコードを紹介するのは恥ずかしいですが、上手いやり方が見つからないのでこんな感じになってます。どなたかシンプルにできる方いませんか…)
d_graph <- data.frame(sample = d_stat$sample,
time = as.character(d_stat$time),
mean.ex = d_stat$mean,
sd.ex = d_stat$sd,
abc = as.character(abc))
この時timeには、as.character()関数を使って文字属性を付与しておきます。(数値としてグラフ描画を行うと、少しおかしなことになります。)
エラーバーの設定
グラフにエラーバーを描画させたいので、aes()関数で、エラーバーに“長さ”という属性を付して、さらにこれをerrorという函に格納しておきます。
errors <- aes(ymin = mean.ex - sd.ex,
ymax = mean.ex + sd.ex)
グラフサイズの設定
後々変更して微調整を簡単にできるように、各種設定を任意の名前をつけた函に代入しておきます。
fig_aspect <- 1
y_max_sd <- c(d_graph$mean.ex + d_graph$sd.ex)
y_max <- max(y_max_sd)
y_axis_max <- c(0, y_max + 0.5) #y軸の長さを、棒グラフの高さに合わせて自動で変えるように設定します。
グラフに表示させるテキストの設定
サンプルや調べる遺伝子の名前は実験ごとに変わるので、編集箇所を集めて作業の効率化を図ります。
sample_column <- c("A", "B")
gene_name <- "PR5"
assay <- "osmostress"
ylab <- expression(paste ("Relative expression level"))
gene_pos_x <- 0.75
gene_pos_y <- (y_axis_max[2] + y_max) * 1/2
gene_nameは、調べたい遺伝子の名前、gene_pos_xとyは、それぞれ、遺伝子の名前を表示させるグラフの座標です。
その他設定
その他、棒グラフの色、フォント、フォントサイズを設定しておきます。
bar_color <- "Greys"
fig_family <- "Arial"
font_size <- 16
5. グラフ作成
早速ggplotでグラフを作成していきます。
g <- ggplot(d_graph, aes(x = sample, y = mean.ex, fill = time)) +
labs(y = ylab, fill = assay) +
geom_bar(position = position_dodge(), stat = "identity", width = 0.7) +
scale_fill_brewer(palette = bar_color) +
geom_errorbar(position = position_dodge(0.7), errors, width = 0.1) +
geom_text(aes(y = y_max_sd, label = sprintf("%s", abc), vjust = -2),
position = position_dodge(0.7), size = 6, family = fig_family) +
scale_x_discrete(limit = sample_column) +
scale_y_continuous(expand = c(0, 0), limits = y_axis_max) +
annotate("text", x = gene_pos_x, y = gene_pos_y,
label = gene_name, family = fig_family, fontface = "italic", size = 6) +
theme_classic() +
theme(legend.title = element_text(size = font_size, family = fig_family),
legend.text = element_text(size = font_size - 2, family = fig_family),
aspect.ratio = fig_aspect,
axis.title.x = element_blank(),
axis.title.y = element_text(size = font_size, family = fig_family),
axis.text.x = element_text(size = font_size, colour = 1, family = fig_family,
angle = 45, hjust = 1),
axis.text.y = element_text(size = font_size, colour = 1, family = fig_family))
plot(g)

plot(g)の出力結果
-
ggplot()
どのデータセット(データフレーム?)を使うのか、縦、横軸に何をセットするかを指定します。 -
geom_bar
グラフの種類によって変更します。geom_lineとかgeom_histogramとか色々あります。 -
scale_fill_brewer()
グラフの色を指定することができます。今回はGreysでシンプルにしてみました。
RNA-seqなどのヒートマップにはpalette = "Spectral"で青〜赤のグラデーションにしてあげるとよいと思います。 -
geom_errorbar
エラーバーの描画に使いました。 -
geom_text
検定結果のアルファベットを表示させるのに使用しました。エラーバーとアルファベットが重ならないように、vjustで位置の指定をします。 -
scale_x_discrete
サンプルの並びを固定するために指定しておきます。(Rはサンプル名をアルファベット順に勝手に並び替えてしまう) -
scale_y_continuous
expand = c(0, 0)を指定することで、グラフの下側をx軸にくっつけることができます。また、limits =を指定して、y軸の高さをいい感じに(エラーバーとか、表示する検定結果がグラフからはみ出さないように)調節してくれます。 -
annotate
グラフ内に、テキストや数式などを表示させることができます。 -
theme()
フォント、フォントサイズの指定など、ディティールを指定できます。論文体裁にしたかったので、フォントはArialで、あとは適当にやりました。
おわりに
今回は、Rを使ってデータの読み込み〜検定〜グラフ出力を行いました。
全ての工程において、なるべく掘り下げて易しく書きました。理解の助けになれば幸いです。もっと説明、プレゼン上手になれるように頑張ります。
実験結果
一応、実験条件についてお話したので、今回の個人的な出力データの解釈についても書いておきます。
ecotype AもBも、ストレス無処理のサンプルに比べて、ストレス処理3日後のサンプルの方でgene Pの発現レベルが有意に増加しており、その増加のレベルは、ecotype AよりもBの方が大きいです。
gene Pは、シロイヌナズナのある特定のストレス応答のマーカー遺伝子として用いられる、ストレス応答シグナリングの下流因子で、この遺伝子の発現上昇はストレスに耐えようとしたかどうかの結果と解釈できます。(遺伝子発現は秒単位で起こるのに、ストレス処理後3日間もおいているのはgene Pがシグナル伝達経路のほぼ最下流だから)
つまり、ecotype A, Bともにストレス応答をしていますが、その程度はBの方がより高いことが示唆される結果となりました。
今回の全R script
シンプルにできる方、どなたかその方法についてご指摘、ご教授くださると嬉しいです。
setwd("~/Desktop/Zenn/bar-graph_2")
library(ggplot2)
library(dplyr)
library(multcomp)
d <- read.csv("sample-time-gene.csv", header = T)
#headerは「sample,time,gene」
#統計検定用のデータ作成
d1 <- mutate(d, sample_time = paste(d$sample, d$time))
d_tukey <- data.frame(sample_time = d1$sample_time, ex = d1$gene)
d_tukey$sample_time <- factor(d_tukey$sample_time)
TukeyHSD(aov(ex ~ sample_time, data = d_tukey))
#検定結果のアルファベット出力
res <- aov(ex ~ sample_time, data = d_tukey)
tuk <- glht(res, linfct = mcp(sample_time = "Tukey"))
mltv <- cld(tuk, decreasing = F)
abc <- mltv[["mcletters"]][["Letters"]]
abc
#グラフ描画用データフレーム作成
d_stat <- d %>%
group_by(sample, time) %>%
summarise_all(list(mean = mean,
sd = sd))
d_graph <- data.frame(sample = d_stat$sample,
time = as.character(d_stat$time),
mean.ex = d_stat$mean,
sd.ex = d_stat$sd,
abc = as.character(abc))
#エラーバー設定
errors <- aes(ymin = mean.ex - sd.ex,
ymax = mean.ex + sd.ex)
#グラフサイズ設定
fig_aspect <- 1
y_max_sd <- c(d_graph$mean.ex + d_graph$sd.ex)
y_max <- max(y_max_sd)
y_axis_max <- c(0, y_max + 0.5)
#figに表示させるテキストの設定
sample_column <- c("A", "B")
gene_name <- "PR5"
ylab <- expression(paste ("Relative expression level"))
gene_pos_x <- 0.75
gene_pos_y <- (y_axis_max[2] + y_max) * 1/2
#その他設定
bar_color <- "Greys"
fig_family <- "Arial"
font_size <- 16
g <- ggplot(d_graph, aes(x = sample, y = mean.ex, fill = time)) +
labs(y = ylab, fill = assay) +
geom_bar(position = position_dodge(), stat = "identity", width = 0.7) +
scale_fill_brewer(palette = bar_color) +
geom_errorbar(position = position_dodge(0.7), errors, width = 0.1) +
geom_text(aes(y = y_max_sd, label = sprintf("%s", abc), vjust = -2),
position = position_dodge(0.7), size = 6, family = fig_family) +
scale_x_discrete(limit = sample_column) +
scale_y_continuous(expand = c(0, 0), limits = y_axis_max) +
annotate("text", x = gene_pos_x, y = gene_pos_y,
label = gene_name, family = fig_family, fontface = "italic", size = 6) +
theme_classic() +
theme(legend.title = element_text(size = font_size, family = fig_family),
legend.text = element_text(size = font_size - 2, family = fig_family),
aspect.ratio = fig_aspect,
axis.title.x = element_blank(),
axis.title.y = element_text(size = font_size, family = fig_family),
axis.text.x = element_text(size = font_size, colour = 1, family = fig_family,
angle = 45, hjust = 1),
axis.text.y = element_text(size = font_size, colour = 1, family = fig_family))
plot(g)
References
たくさん参考にさせていただきました。ありがとうございます。
Discussion
自分はベースとなるグラフテーマ用のR Scriptを作ってフォントの読み込みやテーマ作成をしています。またこのとき
ggplot2::theme_set使ってますね。ただ結構紙面上の都合とかで共通化が難しくて結局グラフ作るところでまたggplot2::themeで一部上書きしたりもしてますね。新しい視点です!まだまだR初心者なので調べながら頑張ってトライしてみます!ありがとうございますm(_ _)m