Snowpark Container ServiceのR言語環境でsingle cell RNA-seq解析をする
*本記事はMediumで投稿されたこちらの記事の日本語版です。
はじめに
snowpark container service(SPCS)が最近すべてのリージョンで使えるようになりました。SPCSは社内アプリケーションのホスト先や、バッチ処理サーバなど様々なビジネスシーンでの利用が可能ですが、コンピューティングリソースをたくさん必要とするゲノム解析や遺伝子データ解析などのバイオインフォマティクスの領域でも応用が可能です。その中でも、single cell RNA-sequencing(scRNA-seq)は、個々の細胞レベルでのRNAの発現をプロファイルする高度な技術であり、多くのコンピューティングリソースを必要とします。今回は10X genomics フォーマットファイルを内部ステージにアップロードし、RパッケージであるSeuratを用いて細胞種ラベリングを行ってみたいと思います。本記事はSPCS上にRstudio Serverのセットアップからはじめていきたいと思います。
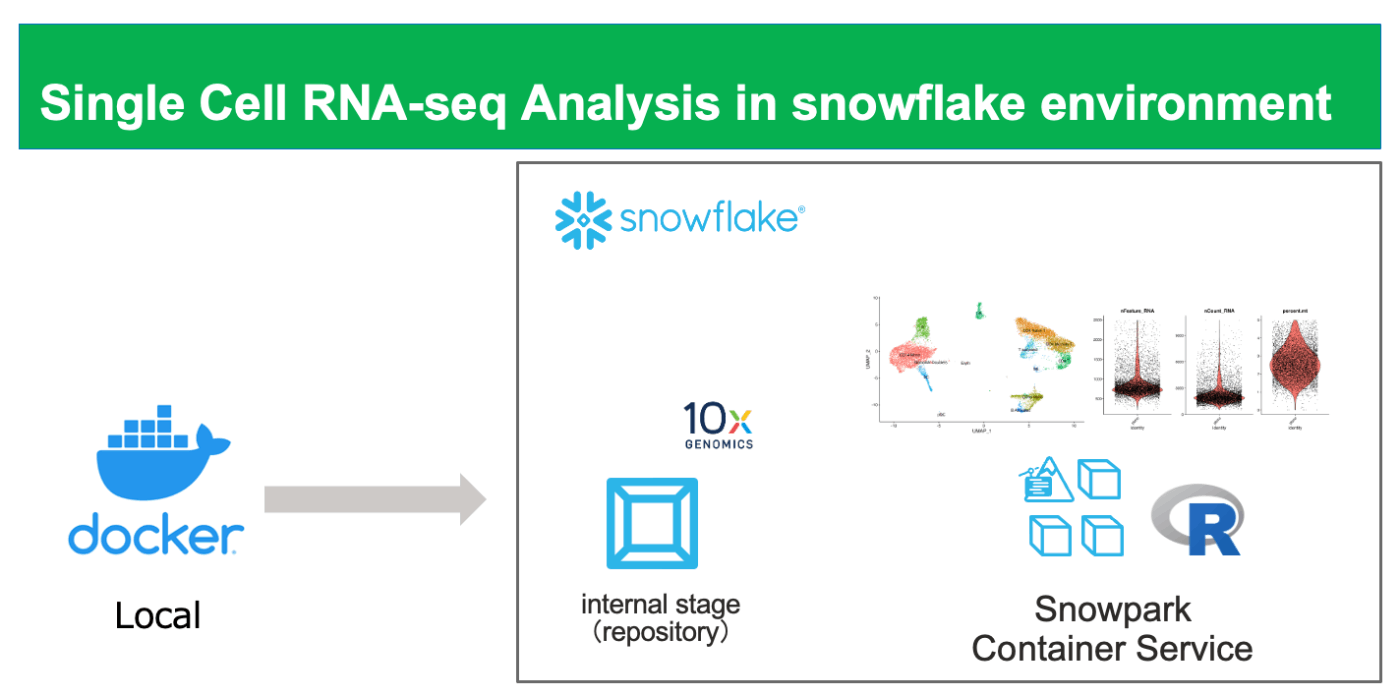
10X Genomic Data formatを解析する構成図
なぜRなのか
Rはバイオインフォマティクス、エピデミオロジー、統計遺伝学など、バイオ系やヘルスケア業界で広く利用されています。またアップデートも頻繁に行われております。データサイエンティスト協会の出している図の中でも、Pythonと並んでRが紹介されています。データエンジニアリングにおけるPythonとRの各言語の強みは以下のとおりです。
Pythonの強み
・AIシステム、ウェブアプリケーション、IoTデバイス、クラウドプラットフォームとの互換性
・機械学習や深層学習モデルを簡単に構築できる
Rの強み
・統計理論に基づいた結果(例:信頼区間、p値)の簡単な計算
・グラフが視覚的に魅力的でシンプル(個人的見解)

PythonとRそれぞれの強み
個人的には以下のような点でR言語を気に入っています。
・バイオインフォマティクスに特化した専門的なライブラリが豊富
・ggplot2による作画がきれい(下図)
・統計解析に強み
・dplyrによるデータフレーム操作が簡便

James Ding et al., 2020 Characterisation of CD4+ T-cell subtypes using single cell RNA sequencing and the impact of cell number and sequencing depth
Snowflake社の技術は主にPythonを使用していますが、これまでSnowflakeでRを使う方法は限られていました。しかし、Snowpark Container Serviceの登場により、Rを使用する可能性が広がりました。Snowflake社が提供するSPCSの画像でも「R」のマークが確認できます。

Rマークが確認できるSPCS
Rstudio Server と Spec
RStudioは、Posit社が管理しているRプログラミング言語を使用するデータ分析者や研究者が開発するための統合開発環境(IDE)です。コードの編集、実行、デバッグ、可視化など、Rプログラムの全ての側面を支援します。またRstudioをがLinux上のWebサーバーをベースに動かすためにRstudio Serverというソフトも開発されており、ブラウザからRstudioの利用が可能です。
RockerプロジェクトのDockerコンテナ
Rockerプロジェクトでは、R言語を中心とした様々なDockerイメージが提供されています。各イメージは特定のニーズに応じて設計されており、以下のように階層化されています。
r-ver
- 内容: Rがインストールされたイメージです。バージョンごとにタグが振られています。
- Dockerfile: r-ver:latestのDockerfile
rstudio
-
内容:
r-verの上にRStudioをインストールしたイメージです。 - Dockerfile: rstudio:latestのDockerfile
tidyverse
-
内容:
rstudioの上にtidyverseパッケージやdevtoolsパッケージなどをインストールしたイメージです。 - Dockerfile: tidyverse:latestのDockerfile
verse
-
内容:
tidyverseの上にTinyTeXなどをインストールしたイメージです。 - Dockerfile: verse:latestのDockerfile
geospatial
-
内容:
verseの上に各種Rパッケージをインストールしたイメージです。地理空間分析に特化しています。 - Dockerfile: geospatial:latestのDockerfile
今回はRockerプロジェクトの公開しているimageをベースにSPCS上にホストしていきます。
また今回は私の方で、10X genomics フォーマットファイルのsingle cell RNA-seqデータを解析するためのライブラリ(Seurat)をインストール済みのimageを作成しました。docker pull kinngut/single-cell:latest で使えます
パッケージインストールをする作業をRstudioから行うには、SPCSが外部のインターネットに出る必要があります。そのためネットワークルールを定義してEXTERNAL ACCESS INTEGRATIONといった操作が必要になりますので、ネットワークルールを定義してEXTERNAL ACCESS INTEGRATIONといった操作を行います。
USE ROLE ACCOUNTADMIN;
CREATE NETWORK RULE allow_all_rule
TYPE = 'HOST_PORT'
MODE= 'EGRESS'
VALUE_LIST = ('0.0.0.0:443','0.0.0.0:80')
;
CREATE EXTERNAL ACCESS INTEGRATION all_access_integration
ALLOWED_NETWORK_RULES = (allow_all_rule)
ENABLED = true
;
ネットワークルールとEXTERNAL ACCESS INTEGRATIONを作成したら、specファイルに追記し、サービスを作成します。これで問題なくパッケージインストールができるようになります。
CREATE SERVICE singlecell
IN COMPUTE POOL tutorial_compute_pool
FROM SPECIFICATION $$
spec:
containers:
- name: singlecell
image: <registry_hostname>/tutorial_db/data_schema/tutorial_repository/kinngut/single-cell:latest
env:
DISABLE_AUTH: true
volumeMounts:
- name: rstudio
mountPath: /rstudio/data
endpoints:
- name: rstudioendpoint
port: 8787
public: true
volumes:
- name: rstudio
source: "@TUTORIAL_DB.DATA_SCHEMA.tutorial_STAGE"
uid: 0
gid: 0
$$
EXTERNAL_ACCESS_INTEGRATIONS = (ALL_ACCESS_INTEGRATION)
MIN_INSTANCES=1
MAX_INSTANCES=1;
これにより、ingress_urlにアクセスするとRstudioが起動できます。

Rstudioが起動した様子
10X genomicsフォーマットの解説+内部ステージにデータのアップロード
10x Genomics社は、シングルセルRNAシーケンシング技術を提供する企業で、低コストで大量の細胞を分析することができるため、研究者に広く普及しています。10x Genomics社のものは独自のフォーマットで作成されています。10XGenome社の独自フォーマットを解析するために、cellrangerという特別なライブラリを提供していますが、こちらはSeuratでも解析可能ですので、今回はSeuratで解析します。
10x Genomics社独自フォーマットのデータは以下のようなファイル構成となっております。
$ tree filtered_feature_bc_matrix
filtered_feature_bc_matrix
├── barcodes.tsv.gz
├── features.tsv.gz (またはgenes.tsv.gz)
└── matrix.mtx.gz

Stephanie Hicks「Welcome to the World of Single-Cell RNA-Sequencing」
それではsnowflakeの外部ステージにアップロードして、SPCS上のRstudioで解析してみます。データは250MB以下ならsnowsight上から内部ステージにアップロードできます(それ以上はsnowSQLのPUT経由でいれる)ので比較的軽量なbarcodes.tsv.gzとfeatures.tsv.gzファイルはsnowsightのUI経由でアップロードしてみます。matrix.mtx.gzの容量が250MB以上になる場合は、snowsqlのPUTコマンド経由でアップロードします。

snowsight経由でのファイルアップロード
snowsql -a [アカウント名] -u [ユーザー名]
PUT file://[ローカルのファイルパス] @[ステージへのパス];

予めspecで設定したステージにファイルをアップロードすると、Rstudioのターミナルでボリュームに設定したパスの位置にファイルが確認できるようになります。
解析に用いるコード
今回用いるデータセットは以下になります。
論文名: Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19
データセット:GSE149689
この論文では重症COVID-19の炎症進行要因でタイプIインターフェロン応答の役割についてSingle Cell RNA-seq解析を用いています。またこの論文で用いられたGSE149689は、下記の論文で再解析されています。
Early peripheral blood MCEMP1 and HLA-DRA expression predicts COVID-19 prognosis
この論文では重症COVID-19の予後バイオマーカー: CD14+細胞のMCEMP1およびHLA-DRA遺伝子発現を見るために、GSE149689を含む7つのデータセットをあわせて再解析しています。今回は、Early peripheral blood MCEMP1 and HLA-DRA expression predicts COVID-19 prognosisにあるFig4bを、SeuratでGSE149689データセットを解析することで再現してみましょう。完全に同じにすることは難しいですが、似たFigは作ることができます。

解析に使うコードは以下の通りです。
library(dplyr)
library(Seurat)
library(patchwork)
library(SingleR)
library(celldex)
library(SingleCellExperiment)
# Load the dataset
pbmc.data <- Read10X(data.dir = ".")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
# Calculate percent mitochondrial genes
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# Quality control
pbmc <- subset(pbmc, nFeature_RNA >= 200 & nFeature_RNA <= 5000 & percent.mt < 15)
pbmc <- pbmc %>%
NormalizeData() %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData(features = VariableFeatures(object = pbmc)) %>%
RunPCA() %>%
RunUMAP(dims = 1:20) %>%
FindNeighbors(dims = 1:10) %>%
FindClusters(resolution = 0.5)
# Visualization
DimPlot(pbmc, reduction = "umap", label = T)
# Load reference data from celldex
ref <- celldex::HumanPrimaryCellAtlasData()
# Run SingleR to infer cell types of pbmc dataset using reference data
results <- SingleR(test = as.SingleCellExperiment(pbmc), ref = ref, labels = ref$label.main)
#Add inferred cell type labels to pbmc object
pbmc$singlr_labels <- results$labels
#Visualize cell types in a UMAP plot with labels
DimPlot(pbmc, reduction = 'umap', group.by = 'singlr_labels', label = TRUE)
色々と書いていますが、前処理でクオリティの低いcellを除いた後にスケーリングさせ、PCAで次元圧縮させたものをreferenceデータベースを基に細胞種ラベリングを行い、UMAPで可視化させています。
DimPlotにUMAPの結果が可視化できます。

SingleRパッケージによるcell ラベリングを行うと以下のような免疫細胞種ラベリングがされたUMAPを手に入れることができます。

SPCS上で解析された10X genomics フォーマットのscRNA-seqデータ
Snowpark Container Service上で解析をするメリット
Snowflakeにおけるコンピューティングリソースの変更は非常に簡単で、CREATE COMPUTE コマンド時に INSTANCE_FAMILY を変更するだけで対応可能です。これにより、個人でも迅速にクラウドベースの解析環境を設定できるため、大変便利です。特に、リソースを多く必要とする分析環境を求める際に役立ちます。
CREATE COMPUTE POOL tutorial_compute_pool
MIN_NODES = 1
MAX_NODES = 1
INSTANCE_FAMILY = CPU_X64_XS;
INSTANCE_FAMILYを変更するだけでコンピューティングリソースの変更が可能。
Snowflakeは、独自のOAuth認証を提供しており、これを使用することでユーザーごとにコンテナへのアクセスを管理できます。また、簡単なアプリケーションをホスティングする際には、Snowflakeユーザーを作成するだけでログイン認証システムを簡単に構築できます。これは、アプリケーション開発やデータ操作をPython以外の多様なプログラミング言語で行いたい場合にも非常に有効です。
このように、Snowflakeはユーザーにとって柔軟かつ強力なクラウド解析環境を提供し、コンピューティングリソースの変更から認証システムの構築、多言語での開発まで、幅広いニーズに応えることができます。
検証結果
今回はバイオインフォマティクスのscRNA-seq技術に関してSnowpark Container Serviceを用いて解説を行いました。今回はリードカウント済みの10X genomics formatから解析を行いましたが、SPCS上でCell Rangerをホストすればfastqファイルからデータ解析までsnowflake上で一気通貫な解析が実現可能です。次回の検証項目としたいと思います。SPCSを用いた分析環境はバイオインフォマティクスのみならず、様々な研究分野に革新的な解析環境をもたらすと思います。
Discussion