pyGenomeVizでゲノムの比較結果を可視化(visualize synteny patterns using pyGenomeViz)
はじめに
Genbank形式のアノテーションのついたファイルを使ってゲノムの比較結果を可視化してみます。
pyGenomeVizをGUIで使用する例です。
動作確認 (Hardware)
-MacBook Pro
-チップ Apple M1 (Rosetta2 インストール済み)
-macOS Sonoma 14.7.5
-メモリ 16GB
動作確認 (Software)
-Python 3.13.3
-pyGenomeViz 1.5.0
-streamlit 1.44.1
-blast 2.16.0
-mummer 4.0.1
-mmseqs2 17.b804f
手順
1.データの用意
Nature Scientific Reportsに掲載の、こちらの文献を例にとります。
Comparative genomic analysis of Colistin resistant Escherichia coli isolated from pigs, a human and wastewater on colistin withdrawn pig farm | Scientific Reports
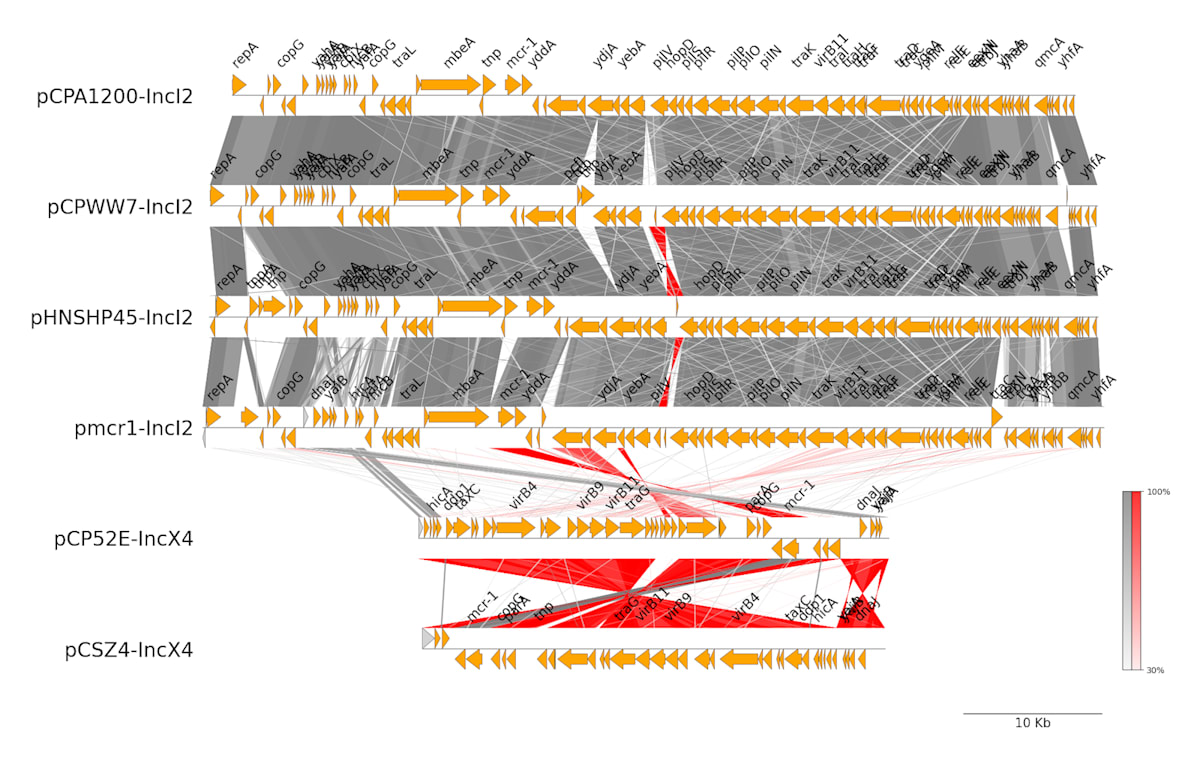
こちらの文献のFigure 3に相当する図を描いてみます。
1-1. Fastaファイルの入手
以前の記事に記した方法で、計6つのプラスミドのfastaファイルをダウンロードしました。
1-2. アノテーションを付与
アノテーションに使用したデータベースのバージョンなどの条件を各株で揃える目的で、自前でアノテーションを付与し直します。
今回は大腸菌(バクテリア)のプラスミドなので、baktaをローカル環境で動かしてアノテーションを付与しました。
baktaを使用すると拡張子が「.gbff」のGenbank形式のファイルが得られました。
2.プログラムのインストール
2-1. pyGenomeVizのインストール
conda(mamba)を使って仮想環境を作成しました。
その仮想環境内に入った後 pyGenomeViz をインストールします。
(※自分の環境ではRosettaを使用しています、ARM64での動作は未検証)
mamba create -n pygenomeviz
conda activate pygenomeviz
mamba install conda-forge::pygenomeviz
2-2. streamlitのインストール
pyGenomeVizをGUI(Web Application)環境で利用するため、
pyGenomeVizの仮想環境に入った状態で streamlit をインストールします。
mamba install conda-forge::streamlit
2-3. Genome Comparison Methodで使うプログラムのインストール
pyGenomeVizでGenome Comparison Methodのメニューで使用する各プログラムをインストールします。
- blast
- mummer
- mmseqs
Blastしか使用しない場合でも、3つとも全てインストールしないとエラーになりました。
pyGenomeVizの仮想環境に入った状態でインストールを実行します。
mamba install bioconda::blast
mamba install bioconda::mummer4
mamba install bioconda::mmseqs2
Release v1.6.1 · moshi4/pyGenomeViz · GitHub
3.描図
pyGenomeVizの仮想環境に入った状態でpyGenomeVizのGUIを立ち上げます。
pgv-gui
ブラウザでpyGenomeViz Streamlit Web Applicationが起動します。
(または「You can now view your Streamlit app in your browser.」の後にターミナルの画面に表示されるURLをブラウザにペーストして実行します)
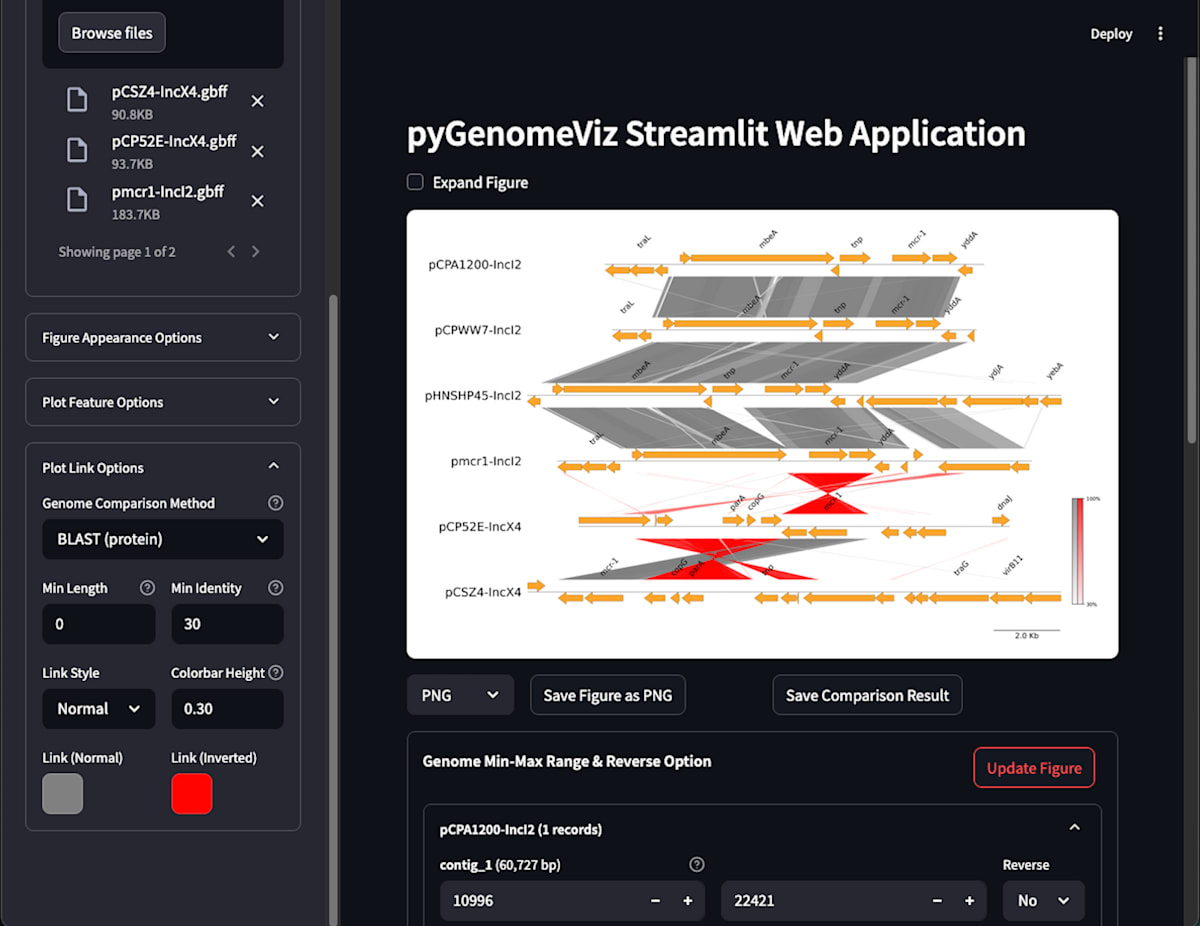
3-1.ファイルの登録
画面左にある「Drag and drop files here」の枠内に描図したいファイルをDrag&Dropして読み込ませます(「Browse files」を押して、目的のファイルを選択することでも可能です)。
ファイルのサイズが100MBまでとのことです。
ファイルを登録すると反映されて描図されます。
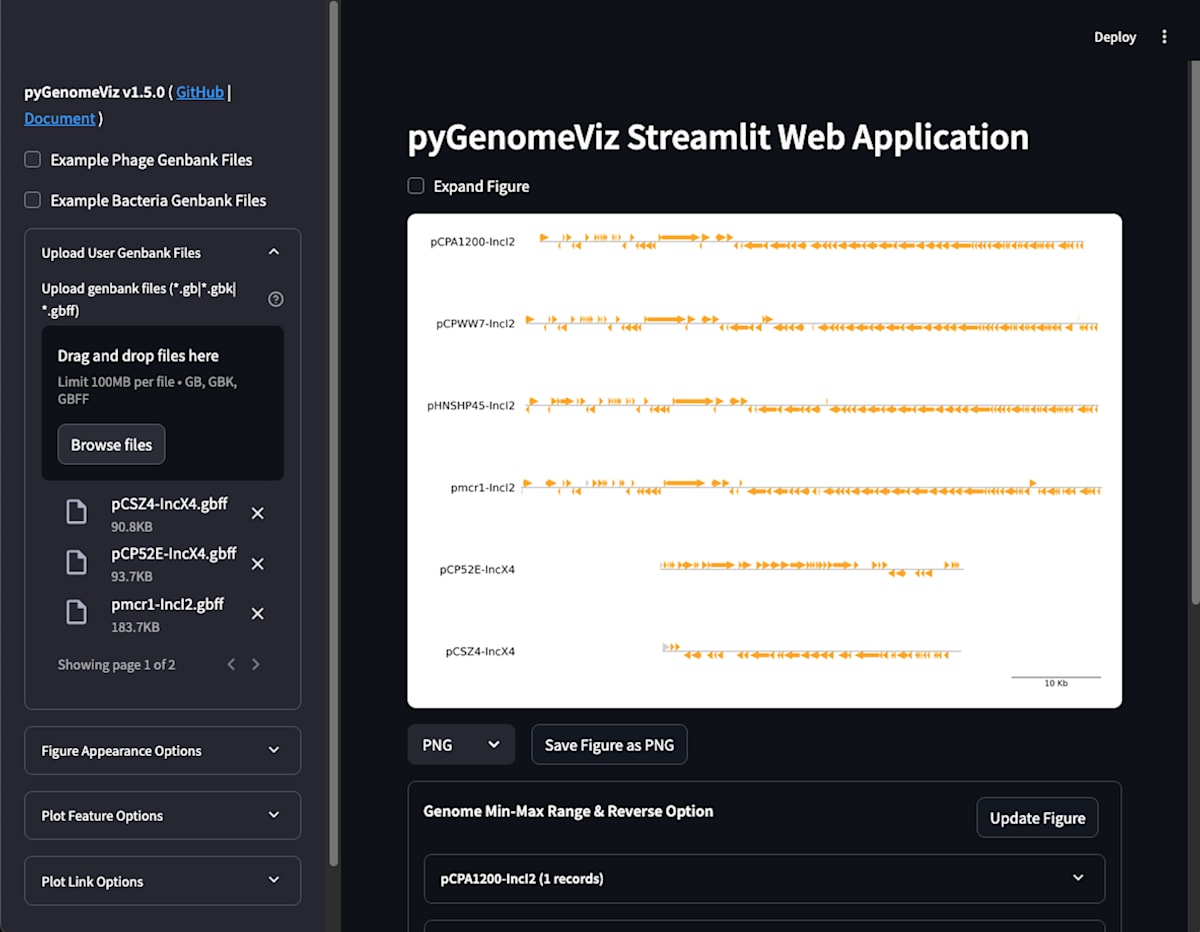
画面上の「Save Figure as PNG」をクリックすると、その時点の画像がpngファイルでダウンロードできます(100dpi)。
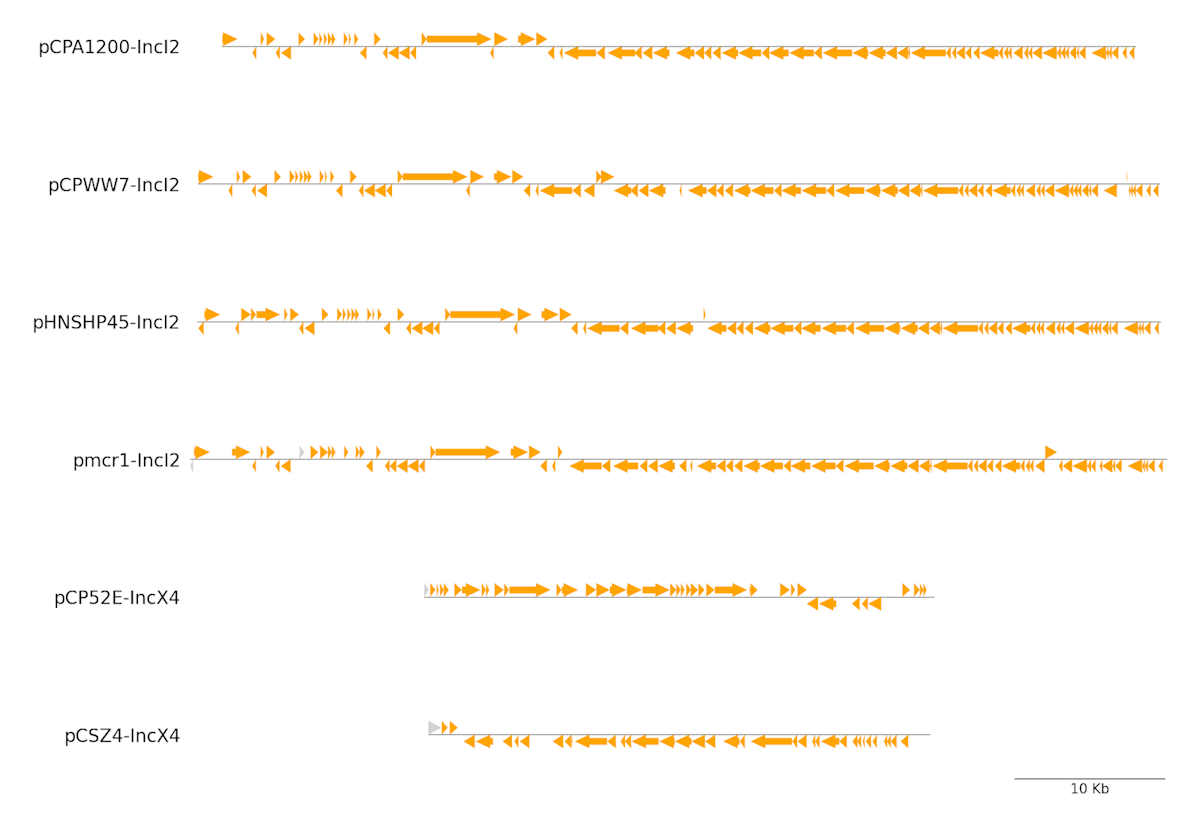
3-2.Figure Appearance Optionsを操作
Figure Appearance Optionsのメニューを開きます。

- Fig Track Height を 1.0 → 0.9(株と株の間を少し縮める)
- Feature Track Ratio を 0.25 → 0.6(featureの図形(今の設定では矢印)を大きくする)
- Label Size を 20 → 24(ラベル名を大きくする)


3-3.Plot Feature Optionsを操作
Plot Feature Optionsのメニューを開きます。

- Plot Feature Types は「CDS」のみ(※複数選択することも可能)
- Plotstyle は「arrow」(矢印)
- Line Width を 0.0 → 0.3(図形に枠線を描く)
- Label Target Track で「All Track」を選択(全ての株でqualifierの情報を表示させる)
- Label type を None → gene(gene qualifierの情報を表示)
- Label Size を 10 → 16(選択したLabel typeのサイズを大きくする)


3-4.Plot Link Optionsを操作
Plot Link Optionsのメニューを開きます。
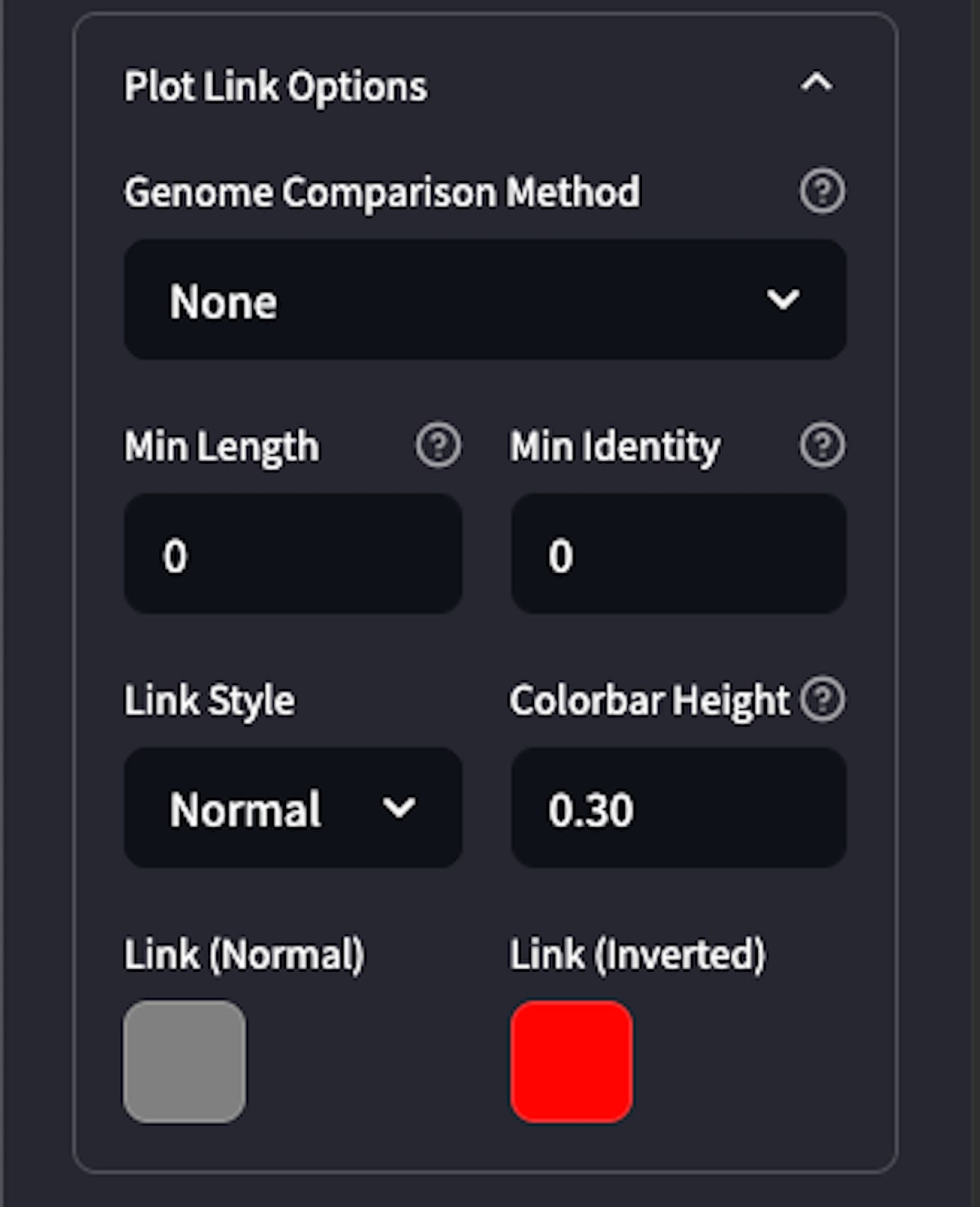
- Genome Comparison Method で「BLAST(protein)」を選択
- Min Identity を 0 → 30
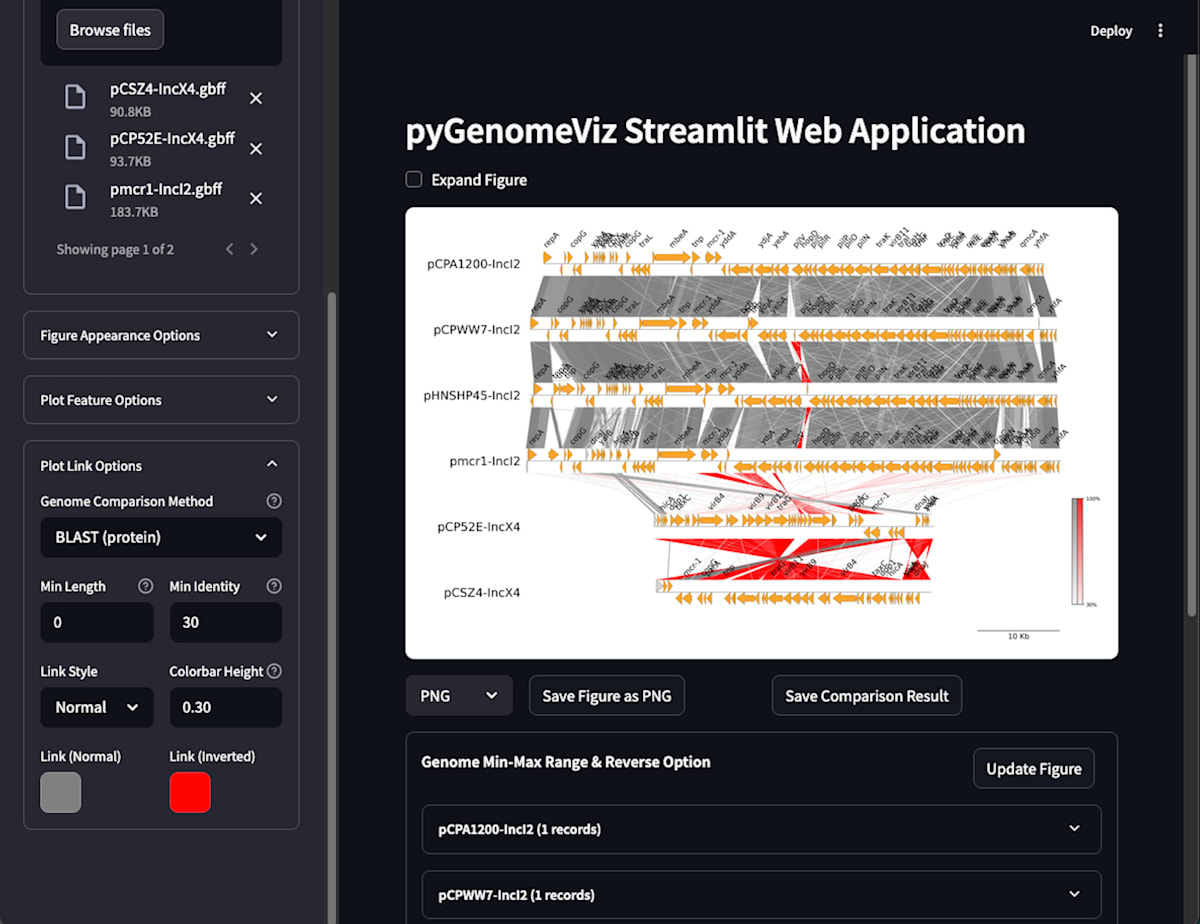
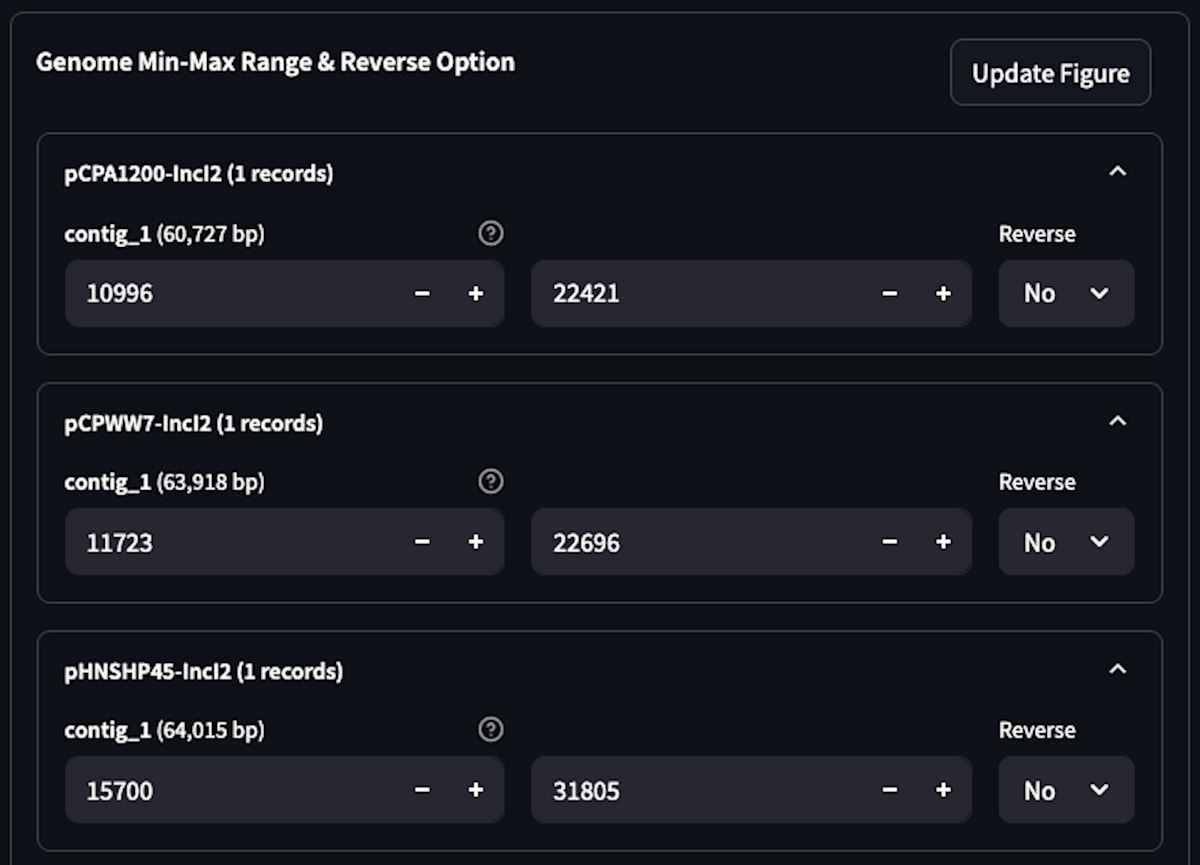
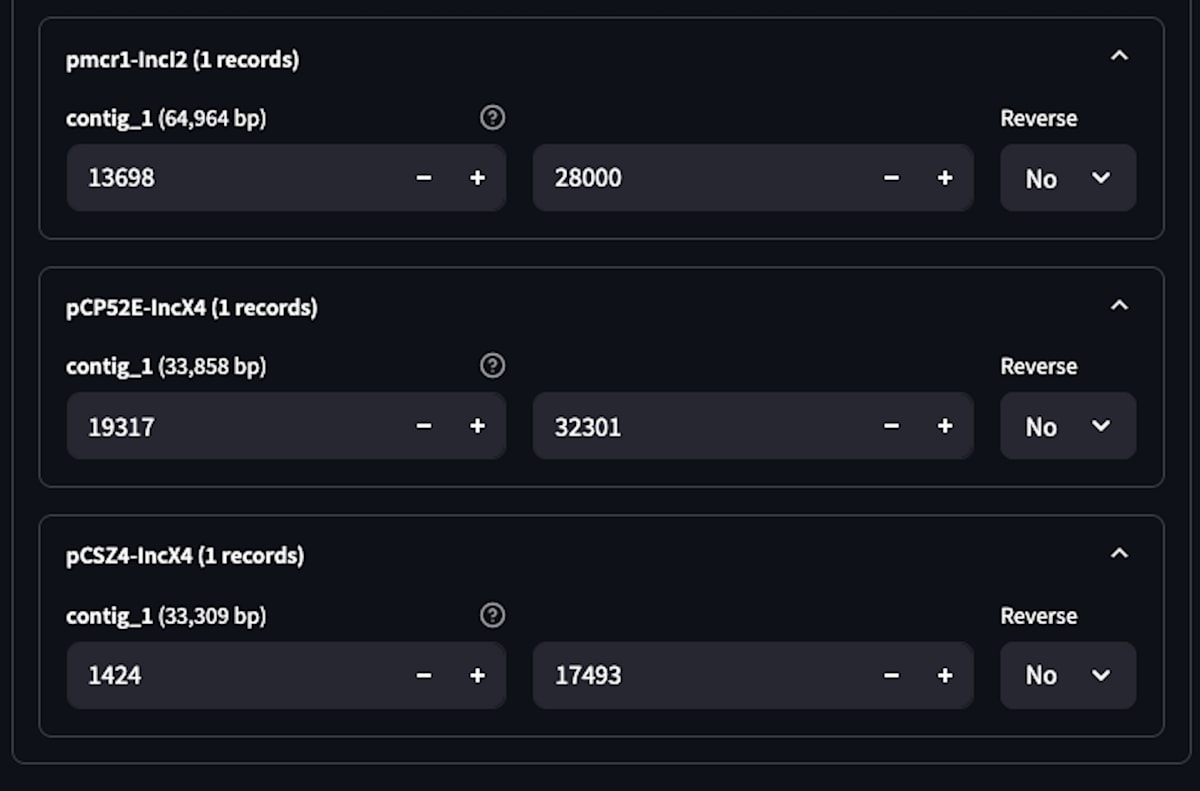
3-5.Genome Min-Max Range & Reverse Optionを操作
画面のGenome Min-Max Range & Reverse Optionの各ファイル名をクリックして開いて、配列中の描図する範囲や配列を反転させるか(Reverse)を指定することができます。
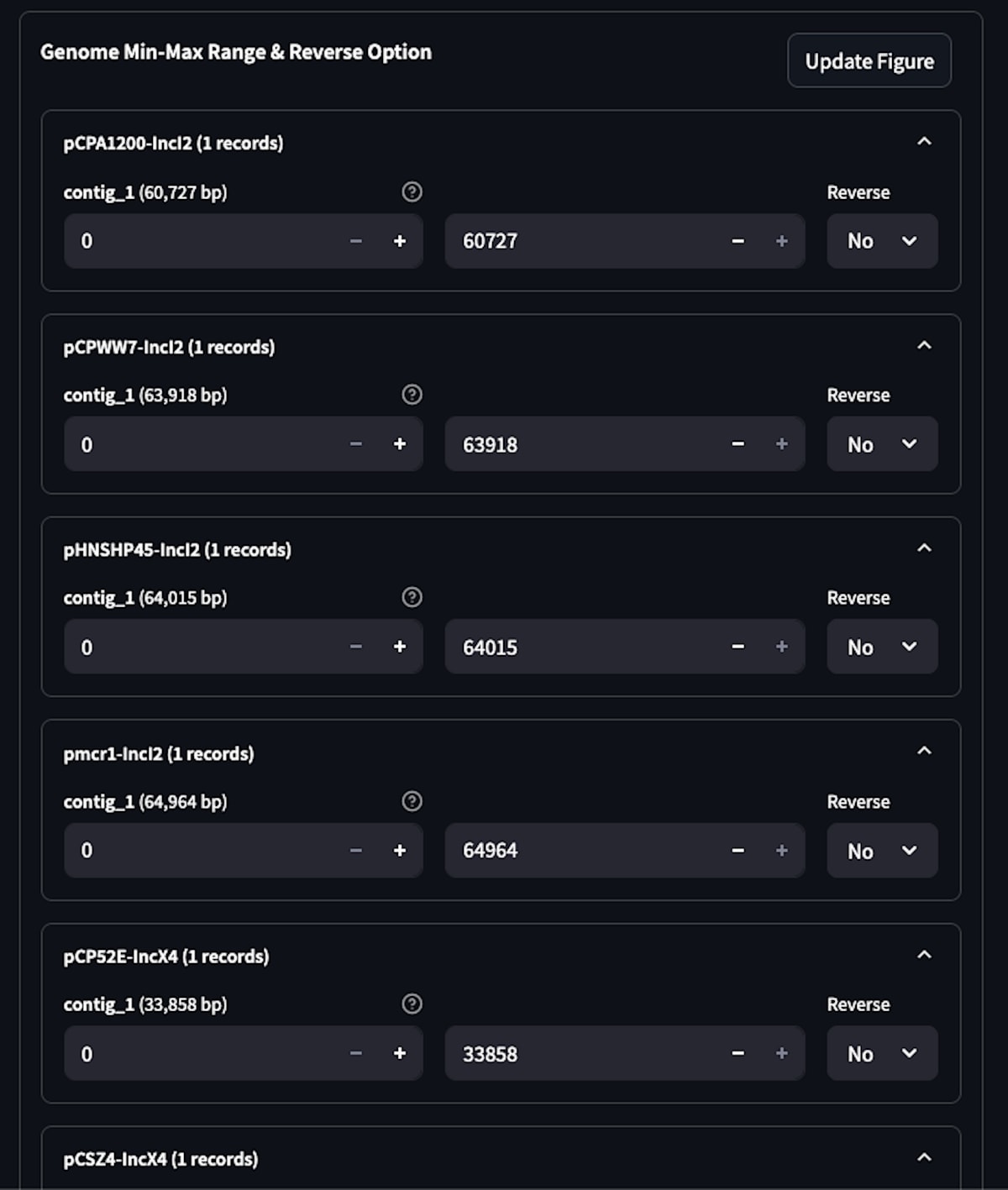
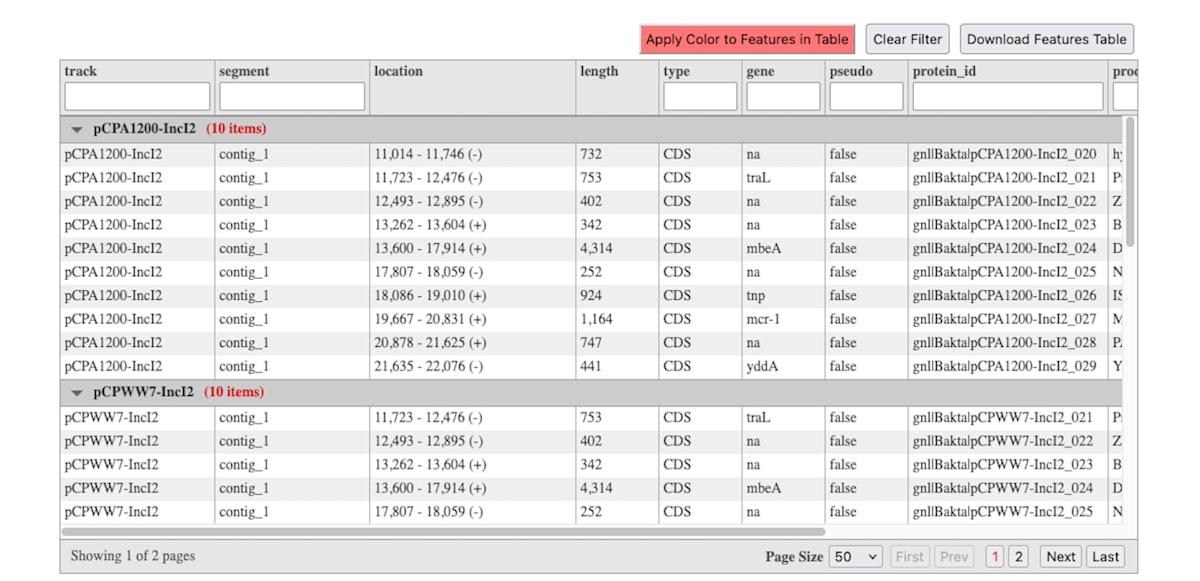
描図する範囲を調べるには、Genbank形式のファイルから直接調べて決めてもよいですし、以前の記事に記した方法で、一旦CDS feature内のqualifierの情報を表形式で取得しておいてその表を見て範囲を絞るのに使う方法もあります。
各々の株の描図する範囲を指定した後「Update Figure」をクリックして更新します。
少し待つと更新されました。
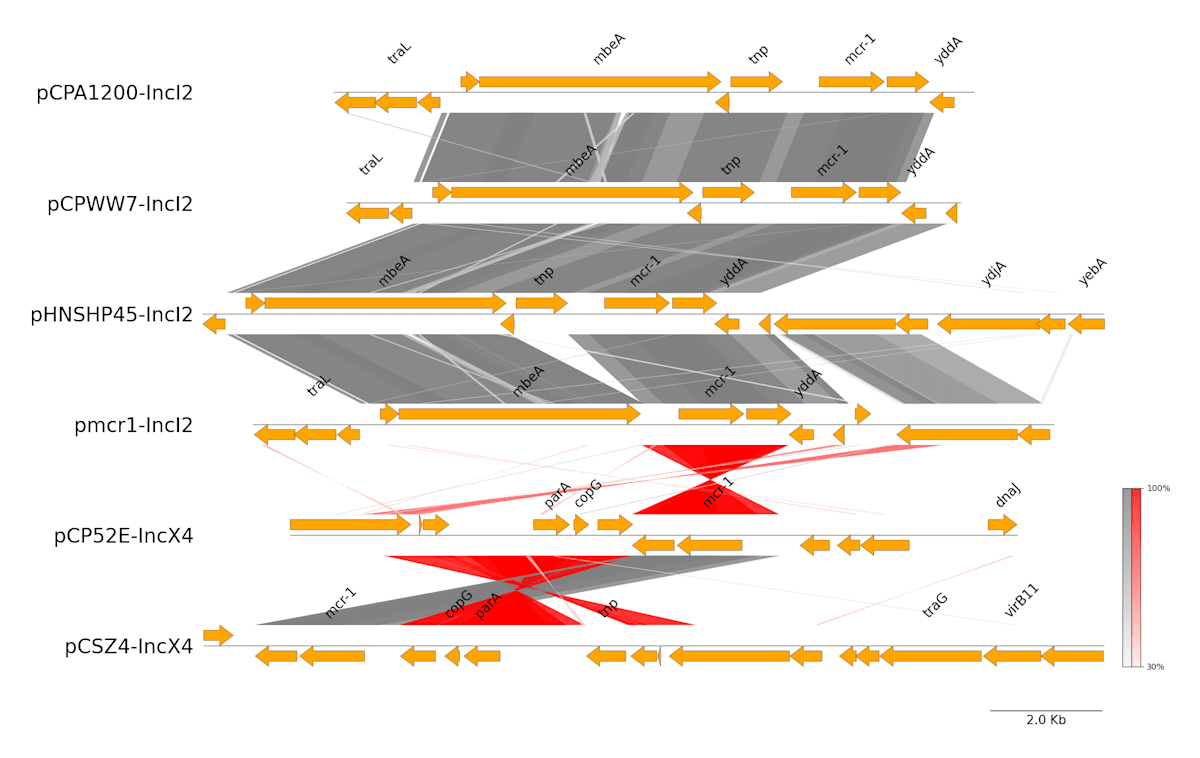
特定のCDSに別の色を指定していきます。
保存形式をPNGから「HTML」に変更してから「Save Figure as HTML」をクリックしてhtmlファイルをダウンロードします。
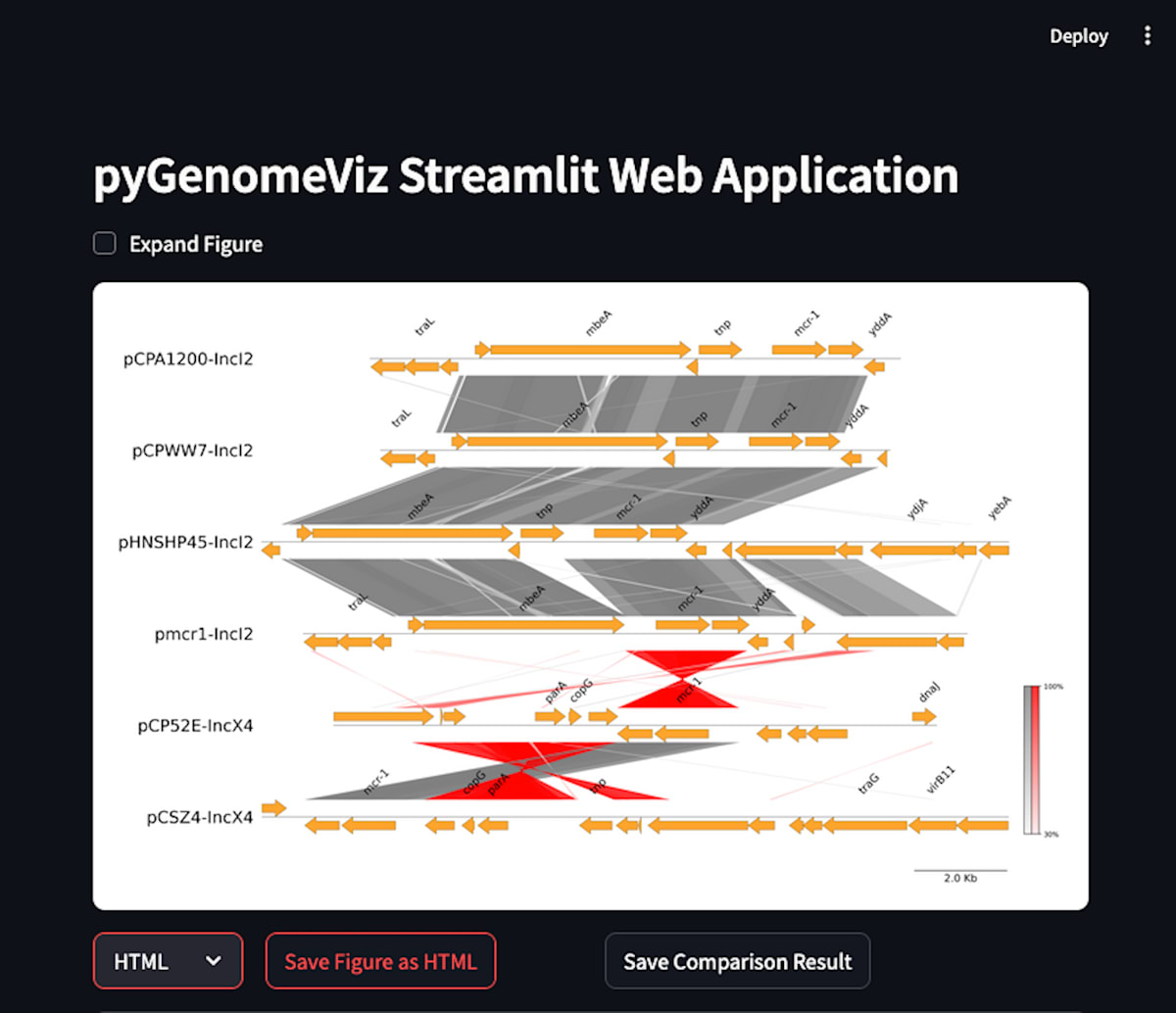
ダウンロードした「pgv_result.html」ファイルをダブルクリックすると、ブラウザでpyGenomeViz Viewerが起動します。
作成中の画像とFeatures Tableが表示されます。

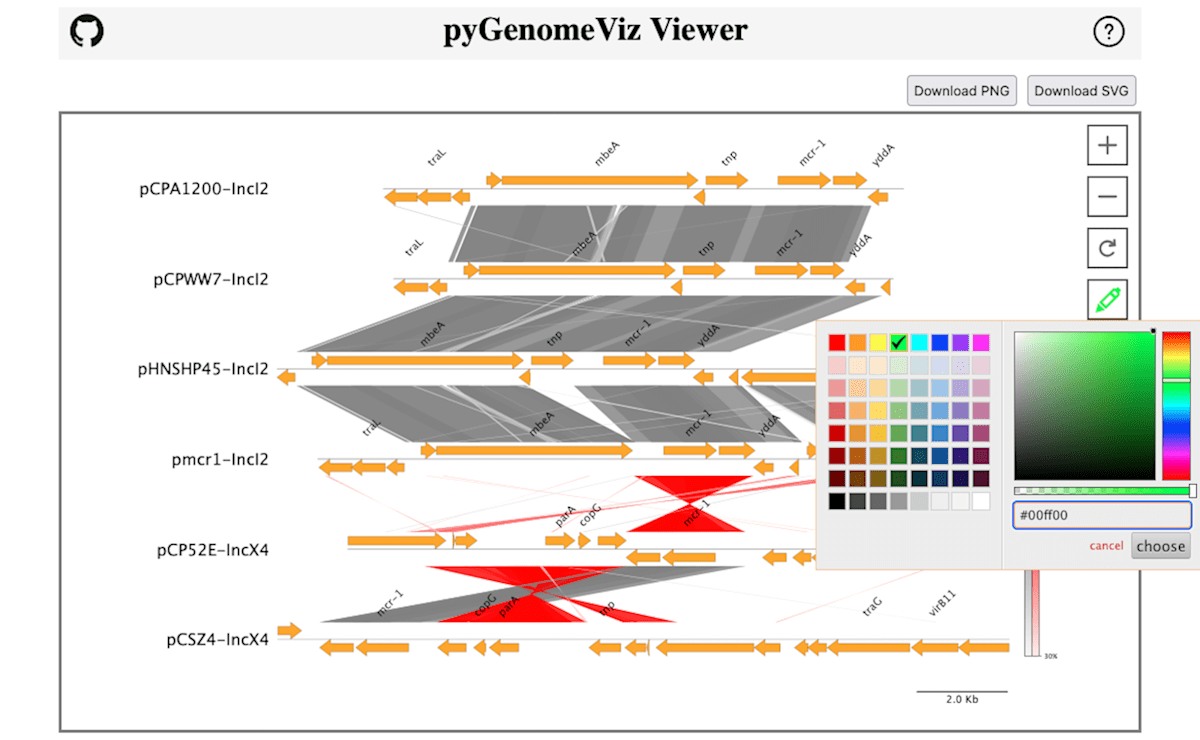
画像の画面右側にある、ペンの形のボタンをクリックするとカラーピッカーが表示されるので色を選択します。
まず Lime(#00ff00)を選択しました。
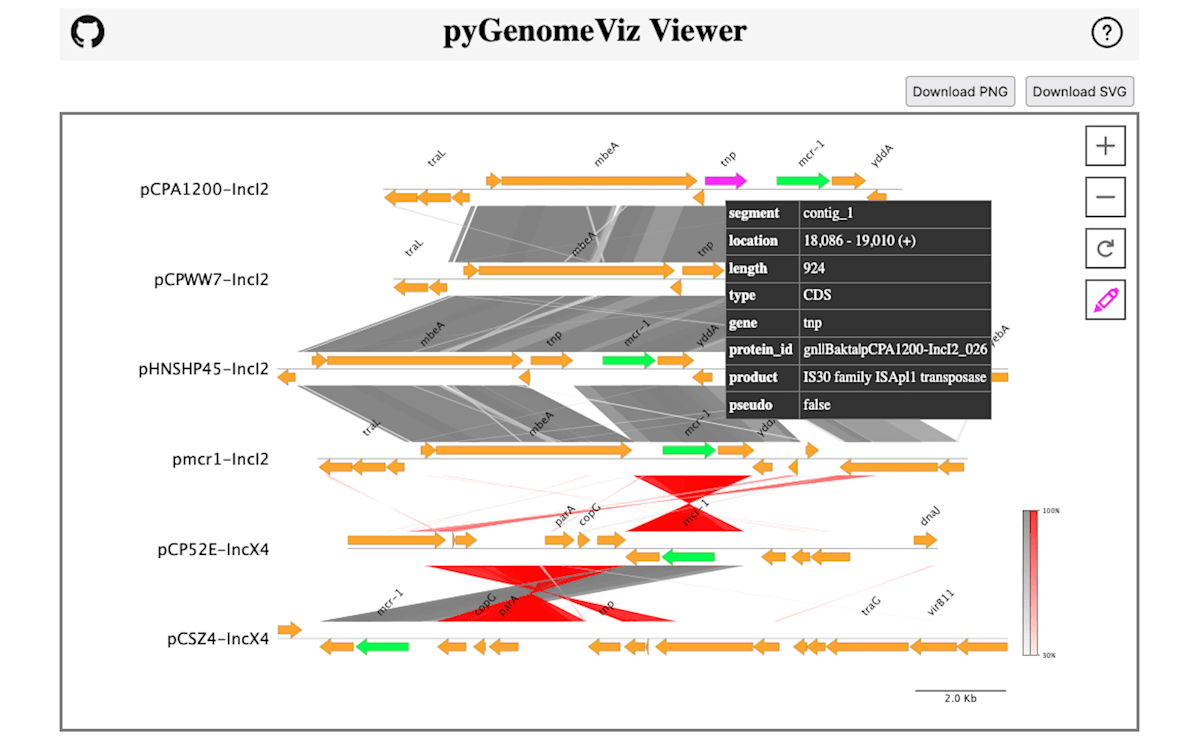
画面上の、色を変えたいCDS featureにマウスのポインターを当てると、そのfeatureの情報がポップアップで表示されます。
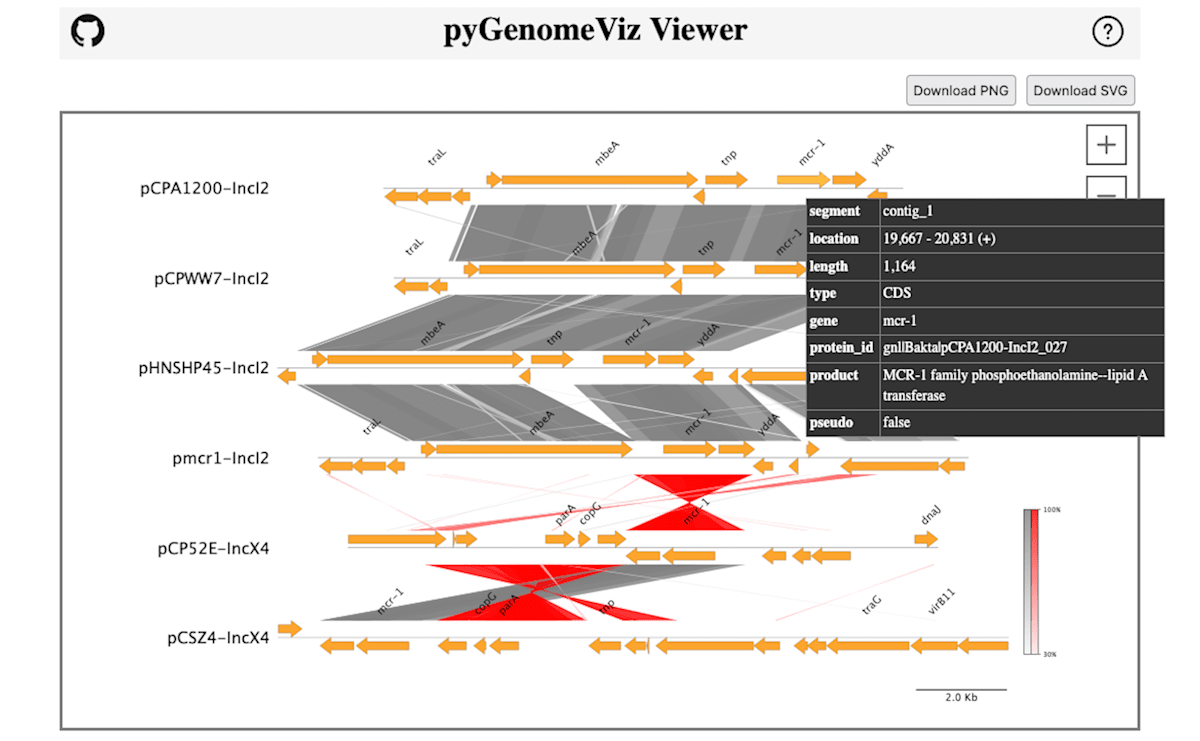
色を変えたいCDS featureの図形上でダブルクリックすると、先程選択した色に変更できます。

各々の株でmcr-1に相当するCDSの色を変更しました。
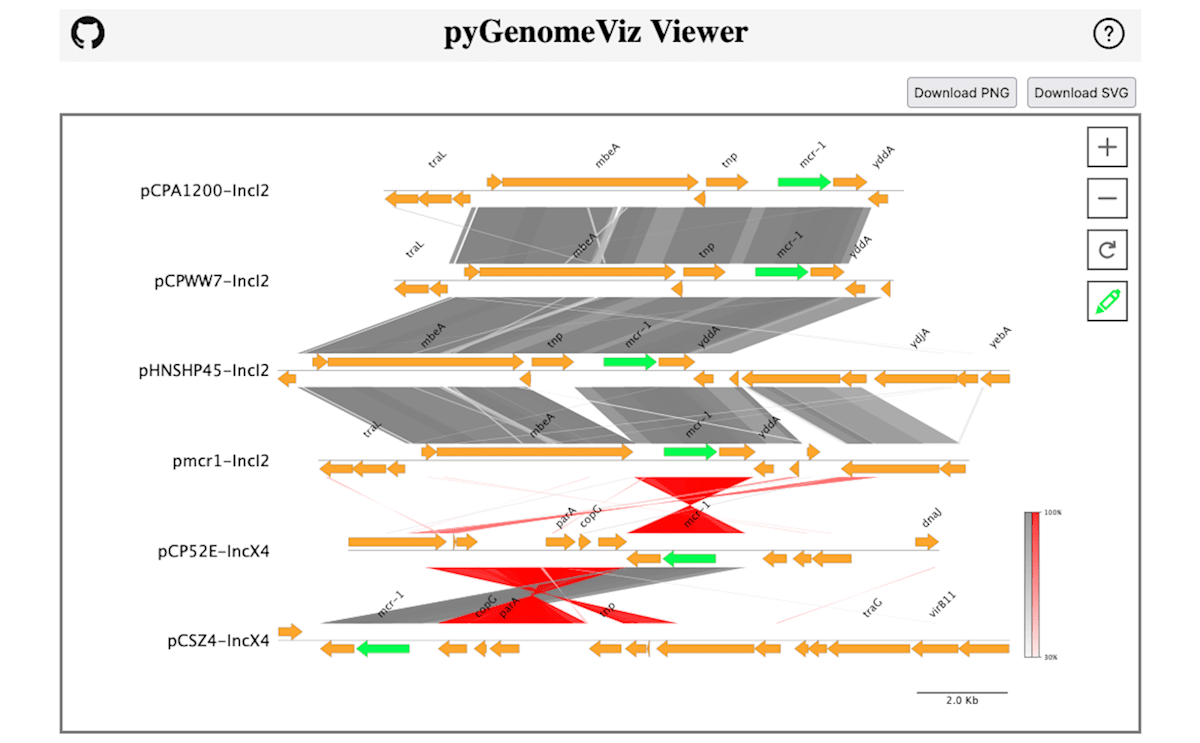
同様の操作で
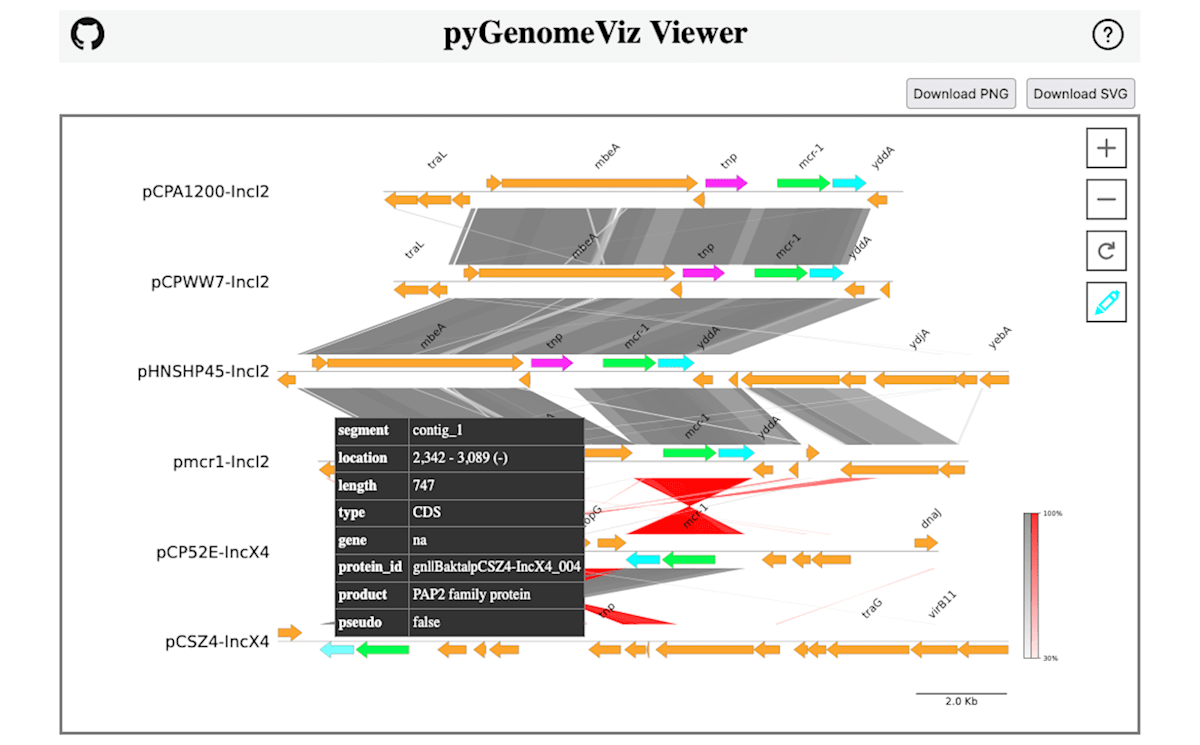
ISApl1に相当するCDSを Fuchsia(#ff00ff)
pap2に相当するCDSを Aqua(#00ffff)
に変更しました。

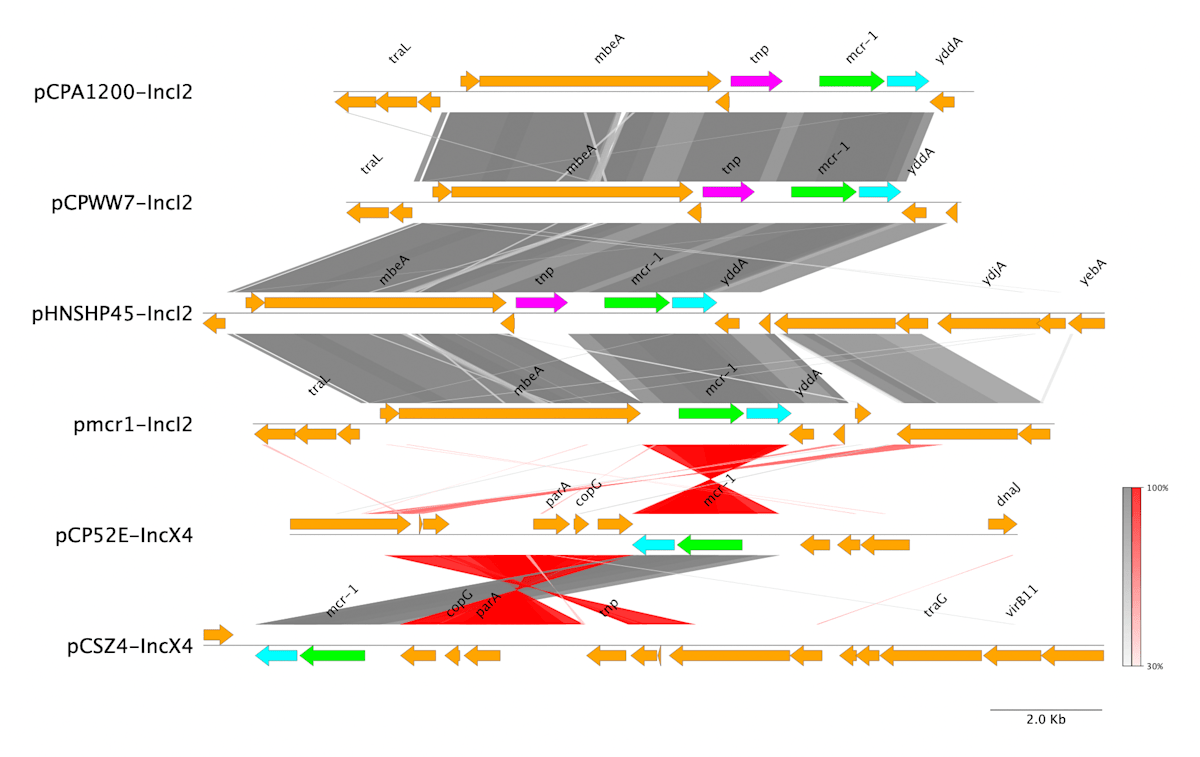
色の指定を終えたらPNG、あるいはSVGで画像をダウンロードして保存します。
作業を終えたらブラウザのpyGenomeViz ViewerおよびpyGenomeViz Streamlit Web Applicationを閉じてCLIに戻って、(control+Cで)Streamlitを終了させて、仮想環境からも出ます。
conda deactivate
おわりに
元の文献の図と比較すると、使用したアノテーションのプログラムやデータベースの違いなどによる部分はありますが、概ね近い図を描くことができたのではないかと思います。
ほんの一例ですが、pyGenomeVizのGUIでの使い方の参考になれば幸いです。
Discussion