お手軽NEB計算 RDKitで分子組み立て
前回 はAvogadroというGUIアプリケーションを使って分子を組み立てました。今回はそれをRDKitでプログラム的に行ってみます。RDKitを使うことで、分子構造や結合の操作を自動化できます(できるとは言っていない)。
今回はRDKitを使ったPythonプログラムがあまりお手軽でないと思います。
対象の反応
こちらのページに紹介されていますフランとアクリル酸メチルの反応です。Endo-とExo-ができる用ですが、今回はRDKitの関数を調べるのが精いっぱいでしたので、なるようになった方で。
RDKit
準備
必要ライブラリ
import numpy as np
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem.rdchem import BondType
from rdkit.Chem.rdForceFieldHelpers import MMFFGetMoleculeForceField
ベクトルの向き合わせ用回転行列
分子を回転させ、結合の向きを合わせる操作がありますので、その準備として2つのnumpyのベクトルの向きを合わせるような回転行列を得る関数を用意します。中身はベクトルの角度からロドリゲスの回転行列を計算するやつで、chatGPTに書いてもらいました。
def rotation_matrix_from_vectors(v1, v0):
"""
v1 から v0 への回転行列を計算する。
v1, v0: 3次元ベクトル(方向ベクトル)
"""
# ベクトルを正規化
v1 = v1 / np.linalg.norm(v1)
v0 = v0 / np.linalg.norm(v0)
# 回転軸(外積)
axis = np.cross(v1, v0)
axis_norm = np.linalg.norm(axis)
# ベクトルが平行または逆平行の場合の処理
if np.isclose(axis_norm, 0):
if np.isclose(np.dot(v1, v0), 1.0): # 同じ方向
return np.eye(3)
else: # 180度回転
# v1 に直交する任意のベクトルを生成
axis = np.cross(v1, np.array([1, 0, 0]))
if np.isclose(np.linalg.norm(axis), 0):
axis = np.cross(v1, np.array([0, 1, 0]))
axis = axis / np.linalg.norm(axis)
return np.eye(3) - 2 * np.outer(axis, axis)
# 回転軸の正規化
axis = axis / axis_norm
# 回転角度
cos_theta = np.dot(v1, v0)
theta = np.arccos(np.clip(cos_theta, -1.0, 1.0))
# ロドリゲス回転公式
K = np.array([[0, -axis[2], axis[1]],
[axis[2], 0, -axis[0]],
[-axis[1], axis[0], 0]])
R = np.eye(3) + np.sin(theta) * K + (1 - np.cos(theta)) * (K @ K)
return R
MM構造最適化
RDKitのMolオブジェクト(三次元構造Embed済)を受け取り、古典力場で構造最適化する関数です。
def optimize_mol(m):
Chem.Kekulize(m)
prop = AllChem.MMFFGetMoleculeProperties(m)
mmff = AllChem.MMFFGetMoleculeForceField(m, prop)
mmff.Minimize()
反応物
ここからDielsAlder反応する分子を作っていきます。
SMILESから分子オブジェクトを作ります。量子化学計算は水素もあらわに扱いますので AddHs も適用します。
furan_smi = "c1ccoc1"
acrylate_smi = "C=CC(OC)=O"
furan = Chem.AddHs(Chem.MolFromSmiles(furan_smi))
acrylate = Chem.AddHs(Chem.MolFromSmiles(acrylate_smi))
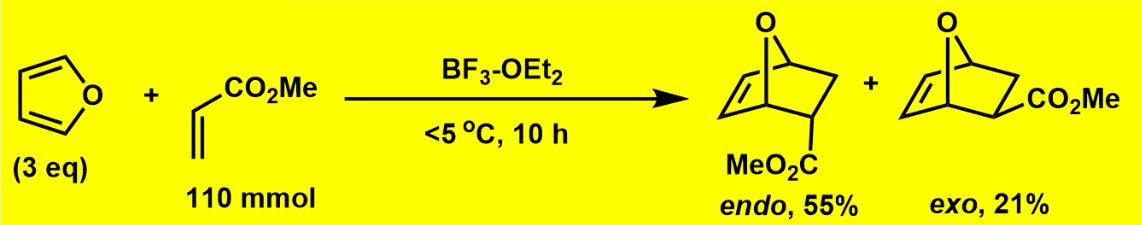
各分子について、各原子に順番に通し番号を振って AtomMapNum に割り当て、画像で出してみて原子の通し番号がどれになったかを確認します。一方でRDKitの各種関数では各原子オブジェクトに振られたindexのほうが使いやすいので、通し番号からindexを見つけるdict mapidx_to_aid も同時に作っておきます。
2D構造式を作る関係で optimizer を使い、 EmbedMolecule で 2D構造式を作ります。
optimizer = AllChem.ETKDG()
count = 1
mapidx_to_aid = {}
for r in [furan, acrylate]:
AllChem.EmbedMolecule(r, optimizer)
for a in r.GetAtoms():
a.SetAtomMapNum(count)
a.SetIntProp("reactant_atom_idx", count)
mapidx_to_aid[count] = a.GetIdx()
count += 1
optimize_mol(r)
Draw.MolToFile(furan, "reactant_furan.png")
Draw.MolToFile(acrylate, "reactant_acrylate.png")
得られた画像は以下のようになります。

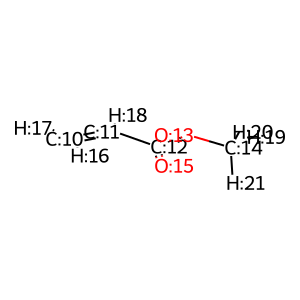
特にアクリル酸メチルの方が、変に立体感が出てわかりにくいですが、 C:10 と C:11 の二重結合が フランの C:3 と C:5 に付加します。
この通し番号をもとに、組み変わる結合をリストにまとめてみます。
newbonds_intermol = [(3, 10), (5, 11)]
bonds_to_s = [(1, 5), (2, 3), (10, 11)] + [(3, 4), (5, 4)]
bonds_to_d = [(1, 2)]
bonds_removed = []
newbonds_intermol は新たに作られる結合の組。 bonds_to_s は2重結合または芳香環結合から単結合になる結合のリストです。二つのリストを結合していますが、前半はDA反応でキーとなる2重結合をリストにしたもので、後半の2つはフランが芳香環として入力されているのでそれを直すためです。 bonds_to_d は2重結合になるものです。今回はフランの酸素の反対側の結合ですね。 bonds_removed は消滅する結合ですが、今回はありません。
反応物3D構造付与
フランとアクリル酸メチルの内部構造に関しては、実は前の節で、for r in [furan, acrylate]: ループ中の optimize_mol(r) で終わっています。ただ、これはそれぞれの分子についてだけに着目して行っていて、これを重ねるとおかしなことになります。

NEB計算がしやすいように移動させます。
反応しやすいように位置合わせ
イメージとしてはこんな感じ。

まずConformerオブジェクト取得。RDKitでは分子の結合情報などを持った Mol オブジェクトに、原子の位置情報を持った Conformer オブジェクトを別に持たせています。原子の位置を見たりいじったりするのならこの Conformer オブジェクトに対して行います。
conf_fran = furan.GetConformer(-1)
conf_acryl = acrylate.GetConformer(-1)
まず移動前のそれぞれの分子の向きを確認。
atom_f3 = conf_fran.GetAtomPosition(mapidx_to_aid[3]) # 通し番号からMolオブジェクト中のidxを得て、furanの3番目の原子の座標をPoint3Dで得る
atom_f5 = conf_fran.GetAtomPosition(mapidx_to_aid[5]) # furan 3番目の原子の座標の Point3D
atom_a10 = conf_acryl.GetAtomPosition(mapidx_to_aid[10]) # アクリル酸メチル原子10の座標の Point3D
atom_a11 = conf_acryl.GetAtomPosition(mapidx_to_aid[11]) # アクリル酸メチル原子11の座標の Point3D
furan_direction = np.array(atom_f5 - atom_f3) # furan C3 -> C5 のベクトル
acrylate_direction = np.array(atom_a11 - atom_a10) # アクリル酸メチル C10 -> C11 のベクトル
実際の回転行列とそれの適用を行います。
R = rotation_matrix_from_vectors(acrylate_direction, furan_direction)
for a in acrylate.GetAtoms():
atompos = conf_acryl.GetAtomPosition(a.GetIdx())
new_atompos = np.array(atompos) @ R.T
conf_acryl.SetAtomPosition(a.GetIdx(), new_atompos)
これが終わったときはこの状態です。

単純に近すぎますので、続いて移動させます。
atom_f3 = conf_fran.GetAtomPosition(mapidx_to_aid[3])
atom_f5 = conf_fran.GetAtomPosition(mapidx_to_aid[5])
atom_a10 = conf_acryl.GetAtomPosition(mapidx_to_aid[10])
atom_a11 = conf_acryl.GetAtomPosition(mapidx_to_aid[11])
center_f = (atom_f5 + atom_f3)/2 # フラン C3とC5 の中点
center_a = (atom_a10 + atom_a11)/2 # アクリル酸メチル C10とC11の中点
shift = np.array(center_f - center_a)
shift[2] = 3.0
for a in acrylate.GetAtoms():
conf_acryl.SetAtomPosition(a.GetIdx(), conf_acryl.GetAtomPosition(a.GetIdx()) + shift)
一度回転して位置が変わったので原子の座標 atom_** を改めて取得し直しています。シフト量の 3.0 は適当です。
これが終わった段階で反応物はこんな感じになります。
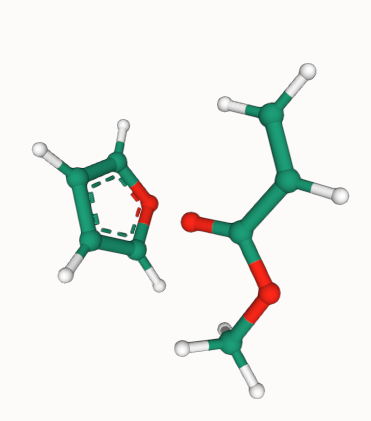
ちょっとわかりにくいですが、フランの C:3 C:5 の方向とアクリル酸メチルのCC二重結合が平行になり、その中心で並行位置も揃わせることができました。
本気でやるなら、さらにそれぞれの分子を平面にフィットさせてその角度を調整したり、それぞれの分子の重心とこれら注目する結合の位置関係からシフト量を算出したりとアルゴリズムはいろいろ考えられると思いますが、いずれにしてもこんな感じで Conformer 経由で三次元座標を取得し、 numpy などを駆使して持っていきたい位置を計算、その結果をまた Conformer を通じてRDKitのオブジェクトに渡して更新するという流れになります。
これ記事を書き始めてから思ったのですが、分子が分かれている反応物系で頑張るのではなく、分子が一つの生成物でまず構造最適化し、その後結合を切って生成物に戻してからまた構造最適化すれば、分子の並進回転を苦労して操作せずとも自然な形でふたつの分子が並ぶんじゃないかなと。まあ今回はRDKitの使い方確認もかねて。
生成物への結合組み換え
product = Chem.RWMol(furan)
product.InsertMol(acrylate)
reactant_set = product.GetMol()
for a in reactant_set.GetAtoms():
mapidx_to_aid[a.GetAtomMapNum()] = a.GetIdx()
これまでの mapidx_to_aid は2つのMolオブジェクト furan acrylate 用になっていたのですが、これからは product の操作を行いますので、 mapidx_to_aid を今度は反応系全体のものに対して見るように更新します。
ここから結合を組み替え、反応を起こします。
- 2重結合になる変化
for a1, a2 in bonds_to_d:
i1 = mapidx_to_aid[a1] # MapIdxをAtomIdxに変換
i2 = mapidx_to_aid[a2]
bond = product.GetBondBetweenAtoms(i1, i2) # 対象のBondオブジェクトを取得
bond.SetBondType(BondType.DOUBLE) # 結合種を二重結合に変更
product.ReplaceBond(bond.GetIdx(), bond) # 変更内容をRWMolオブジェクトに反映
- 単結合になる変化
for a1, a2 in bonds_to_s:
i1 = mapidx_to_aid[a1]
i2 = mapidx_to_aid[a2]
bond = product.GetBondBetweenAtoms(i1, i2)
bond.SetBondType(BondType.SINGLE)
product.ReplaceBond(bond.GetIdx(), bond)
- 切れる結合
for a1, a2 in bonds_removed:
i1 = mapidx_to_aid[a1]
i2 = mapidx_to_aid[a2]
product.RemoveBond(i1, i2)
- 新たにできる結合
for a1, a2 in newbonds_intermol:
i1 = mapidx_to_aid[a1]
i2 = mapidx_to_aid[a2]
product.AddBond(i1, i2, order=BondType.SINGLE)
なんとなく、結合の種類組み換え、切断、生成はこの順でやった方がいい気がします。
最後に構造最適化。 RWMol または EditableMol は原子や結合の変更しかできないので、 Mol オブジェクトにしてからoptimizeします。結合情報が変わっているので、原子の初期位置が反応系でも力場は生成物に適したものになり、最適化の結果生成物の構造が得られます。
product = product.GetMol()
optimize_mol(product)
XYZファイル生成
Chem.MolToXYZFile(product, "product.xyz")
Chem.MolToXYZFile(reactant_set, "reactant_set.xyz")
実は編集前の reactant_set は reactant_set = product.GetMol() のときに新しくConformerが作られたのか、 product の Conformerとは別っぽい。そのため、こんな感じで reactant_set の構造をここで書きだすと、ちょうど結合操作前の構造、つまり反応物系の構造が得られます。
あと、 product.xyz を確認するとendo体ができたようですね。

以上、 この RDKitの節に出てきたコードスニペットを順番に実行、あるいはまとめて一つのpythonスクリプトファイルにして実行してできた xyzファイルをNEB計算で使います。
CP2K
入力ファイル
da.inp に以下の内容を保存します。
&GLOBAL
PRINT_LEVEL LOW
PROJECT diels_alder
RUN_TYPE BAND
&END GLOBAL
&MOTION
&BAND
BAND_TYPE IT-NEB
K_SPRING 0.05
NPROC_REP 1
NUMBER_OF_REPLICA 25
ROTATE_FRAMES T
&CONVERGENCE_CONTROL
MAX_DR 0.004
MAX_FORCE 0.009
RMS_DR 0.002
RMS_FORCE 0.006
&END CONVERGENCE_CONTROL
&CONVERGENCE_INFO
&END CONVERGENCE_INFO
&OPTIMIZE_BAND
OPT_TYPE DIIS
OPTIMIZE_END_POINTS T
&DIIS
MAX_STEPS 200
&END DIIS
&END OPTIMIZE_BAND
&PROGRAM_RUN_INFO
&END PROGRAM_RUN_INFO
&REPLICA
COORD_FILE_NAME ../reactant_set.xyz
&END REPLICA
&REPLICA
COORD_FILE_NAME ../product.xyz
&END REPLICA
&END BAND
&PRINT
&VELOCITIES OFF
&END VELOCITIES
&END PRINT
&END MOTION
&FORCE_EVAL
&DFT
&QS
METHOD PM6
&END QS
&SCF
MAX_SCF 100
# IGNORE_CONVERGENCE_FAILURE
SCF_GUESS ATOMIC
&END SCF
&END DFT
&SUBSYS
&CELL
ABC 30.0 30.0 30.0
PERIODIC NONE
&END CELL
&TOPOLOGY
COORDINATE XYZ
COORD_FILE_NAME ../reactant_set.xyz
&END TOPOLOGY
&END SUBSYS
&END FORCE_EVAL
NEB計算の設定として始点と終点構造から25個の中間ビーズを補間しています。半経験的PM6法を用いて、反応経路上の構造最適化を行います。バンド最適化にはDIIS法を用い、反応物・生成物の構造も同時に緩和されるよう設定しています。
実行
da.inp, reactant_set.xyz, product.xyz を一つのディレクトリに置きます。
cp2k.ssmp da.inp > da.log
95ステップで収束、プログラム実行から終了まで5分41秒でした。
結果可視化用コマンド
前回の記事 でも使いました、CP2Kの出力ファイルから見たい情報を取り出すスクリプトです。前回はbashのワンライナーで書いていたのですが、もうちょっと見やすくpythonスクリプトとして書いておきます。
import matplotlib.pyplot as plt
import numpy as np
data = np.array([float(x) for x in open('diels_alder-1.ener').readlines()[-1].split()[1:] if x.startswith('-')])
plt.plot(range(1, len(data) + 1), (data - min(data))*2625.5, marker='o') plt.xlabel('Bead index')
plt.ylabel('Energy/kJ/mol')
plt.grid(True)
plt.savefig('graph.png')
この python スクリプトは、NEB計算のエネルギー出力ファイル( diels_alder-1.ener )の最終行からビーズごとのエネルギーを読み取り、最小エネルギーからの相対エネルギー(単位:kJ/mol)をプロットします。ビーズインデックス(反応座標上のステップ番号)を横軸、エネルギー差を縦軸に取り、遷移状態を含む反応経路のエネルギープロファイルを描画します。
for file in $(ls diels_alder-pos-Replica_nr_*-1.xyz); do tail -n $(wc -l < ../reactant_set.xyz) $file >> traj.xyz; done
このBashスクリプトは、NEB計算で出力された各ビーズのXYZ形式の構造ファイル( diels_alder-pos-Replica_nr_*-1.xyz )から、それぞれの分子構造部分だけを切り出して、1つのファイル(traj.xyz)に順番に連結します。
tail -n 部分では、各構造ファイルの最後の原子情報(座標部分)だけを取り出しています。行数のカウントには元の反応物構造(reactant_set.xyz)の原子数を利用しています。
作成されたtraj.xyzは、VMDなどの可視化ソフトでアニメーションとして読み込むことで、反応座標に沿った構造変化を動画のように確認することができます。
結果
graph.png はこんなの。

traj.xyz をAvogadroで開き、Animationで動画にするとこんな感じです:

本当はやりたかったこと
化学反応のSMARTS表記を使って、newbonds_intermol bonds_to_s bonds_to_d bonds_removed を自動で作れるようにしたら、結構な化学反応のNEB計算用ファイルがデータベースから自動的に作れるようになる気がします。ですが、RDKitの反応を適用するやつをやってみると、反応テンプレートに該当する原子の AtomMapIdx が消えてしまい、もとの原子の AtomMapIdx を追跡するのが面倒になったので、これは次のステップの話だなと思って止めました。
まとめ
今回は、RDKitを使って分子の3次元構造を構築・編集し、CP2KでNEB計算を行う一連の流れを紹介しました。
NEB計算の精度には、反応物・生成物の構造が重要なのですが、さすがにそれはRDKitは良しなにやってくれません。ただ、「この原子・この結合はこう並べた方がいいんじゃないか」というアイデアがあれば、またそのアイデアをアルゴリズム的に作り出せれば、あとはRDKitで自動化し、自動的にNEB計算用の構造ファイルを生成できるようになります・・・・なればいいな。
Discussion