お手軽NEB計算
概要
NEB計算は化学反応の反応機構を知るのにとても便利です。化学反応の特徴や大まかな活性化エネルギーの把握が簡単な入力(個人差あり)で可能です。遷移状態計算の入力につなげることもできますので、より詳細な計算を行うときの第一歩としても有効です。
NEB計算は分子構造作成に始まり分子構造作成に終わると言います(今思いついた)。そこで分子構造作成の手順を詳し目に、無料で使えるソフトのみでNEB計算を試せる手順を書いておこうと思います。
ソフトウェア
分子構造作成:Avogadro
最新版はバージョン2ですが、事情により1のものを使います。よほど厳しいセキュリティを課している組織でない限り使えると思います。
NEB計算:CP2K
正直Linux上でのビルドがお手軽ではなかったですが、それは別のお話。既にインストールが終わったマシンがあるものとして話を進めます。
(Linuxの使い方の説明を始めると、それだけで終始して計算化学にたどり着けないので・・・)
対象の反応
Claisen転移反応を試します。
とくにWikipediaにあるこちらの反応をやってみます。

(出典:Wikipedia)
ここでは R がメチル基とします。つまり、1-メチルアリルビニルエーテルから4-ヘキセナールへの転位反応を扱います。
分子構造作成:Avogadro
反応物
まずAvogadroを立ち上げます。
ここから分子を入力します。原子を一つ一つ追加していくツールで組み立ててもいいですが、反応物の構造式
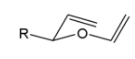
を睨んでいると C=CC(C)OC=C という文字列が浮かんでくると思います(難しければ、PubChemで構造式検索の入力フォームで構造式を入れると対応するSMILESを表示してくれます。また物質名を知っていれば検索してWikipediaかPubChemのページを見つけるとSMILES表記があるかと思います。)
このSMILES表記を使えば一発で入力できます。

メニューから Build → Insert → SMILES... を選択します。すると小窓が表示されます。
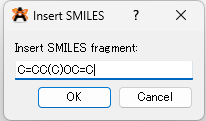
ここに先ほどのSMILES文字列を入力し、OKを押します。

青いものに包まれた分子が出現しました。この青いカバーは原子や結合が選択状態にあることを示しています。 メニューから Select → Select None を選択すると消せます。


なんか直線的に並んでいて変なので、構造最適化してやります。AvogadroはUFFやGAFF力場で構造最適化する機能があります。

上のほうのツールバーから Eと下向きの矢印があるアイコンをクリックします。すると左のパネルに構造最適化に関するメニューが出てきます。

これの一番下の Start をクリックするとリアルタイムで構造最適化されます。
反応物の構造はこれでもいいのですが、遷移構造を見ると舟形、つまり2つの二重結合が程よく近づいている状況がいいようですので、形を変えてやろうとおもいます。
ここから、Avogadroのバージョンを1としたい理由の機能を使います。
構造最適化中、適当な原子をドラックします。するとばねで繋がったビーズを物理演算で動かす感じで、分子をマウスで動かすことができます。
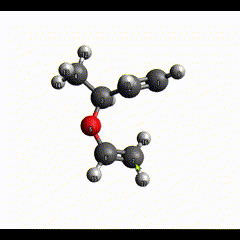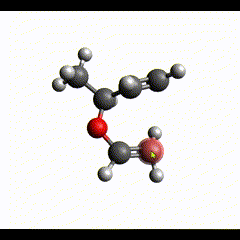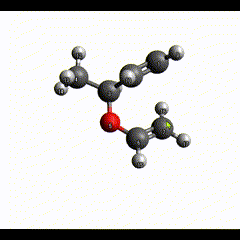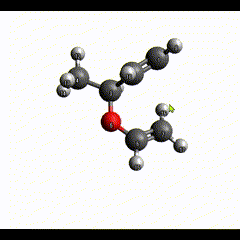
画像中の赤いカバーで包まれている原子が今ドラッグしているところの原子です。いま画像の上の方に引っ張っています。
構造最適化中だと、ドラッグ中の原子に向かって分子が引きずられている様子が見られます。(分子が大きすぎるとフリーズしますが)
さて頑張って(といっても今回のは1分もあればできますが)これくらい近くに持ってきました。

これを保存しておきます。 File → Save As.. を選び、 reactant.xyz というファイル名で保存しておきます。
さて、NEB計算では原子同士の対応が重要です。有機化学から量子化学計算に入ると、油断すると水素の扱いが雑になるのですが、ここを間違えると無茶な結合の組み換えや回転が起こることとなってしまい、計算に失敗してしまいます。一応各原子がどういうラベルがついているかを見ておきます。

左側の操作パネルの、 Display Types から Label にあるチェックボックスにチェックを入れてみます。

すると各原子上に、それぞれ元素ごとに何番目の原子かを示したラベルが付きます。ただこれではXYZファイルの何行目かが分かりにくいので、ちょっと変えてやります。先ほどの Label の行の右側にスパナアイコンがありますが、これをクリックすると表示するラベルを設定できます。

これで Atom number を選んでやると、その原子が分子中で何番目にあるかを示す数字を表示します。

もしXYZファイルを直接編集することがあるなら、この数字を参考にしてみてください。XYZファイルだと最初の行が原子数、次の行がタイトルで、3行目から原子の記録が始まりますので、原子上の数字が N の原子の座標はXYZファイルだと N+2 行目に書いてあります。
生成物
続いて、結合を組み替えて生成物を作ってやります。ここから分子を直接いじれるツールを使います。
注意 : ここからの操作はミスりやすいです。ミスったときは炭素原子が消えたり、余計な炭素原子が増えたりします。AvogadroはUndo機能はありますが、こういうときUndoしてもおかしなことになりがちで、だいたいのケースで一からやり直した方が早いです。つまり、 reactant.xyz を開きなおしてそこから編集しなおします。ここは個人的に気に入っていない部分ですね。
また、ここからのパートでは Ctrl-S を押すとせっかく作った reactant.xyz に上書きされてしまう可能性がありますので、できるだけ Ctrl-S を押さないようにするか、 reactant.xyz を別に作っておきましょう。 コピーして product.xyz とし、これを編集、最後までうまく編集できたら保存するという手順が良いかもしれません。
ツールバーから鉛筆アイコンを選択します。

すると左側の操作パネルに分子編集ツールのオプションが出ますが、 Adjust Hydrogens のチェックを外します(重要)。

基本的にこのツールで結合次数の変更や結合の組み換えを行いますが、もし水素調節が入ると、結合次数に合わせて水素原子の増減がなされ、結果原子の対応が滅茶苦茶になります。
このツール選択時、結合をクリックすると 単結合→二重結合→三重結合→単結合 ・・・ と変更できます。まず右下の二重結合を三重結合にしたところです。
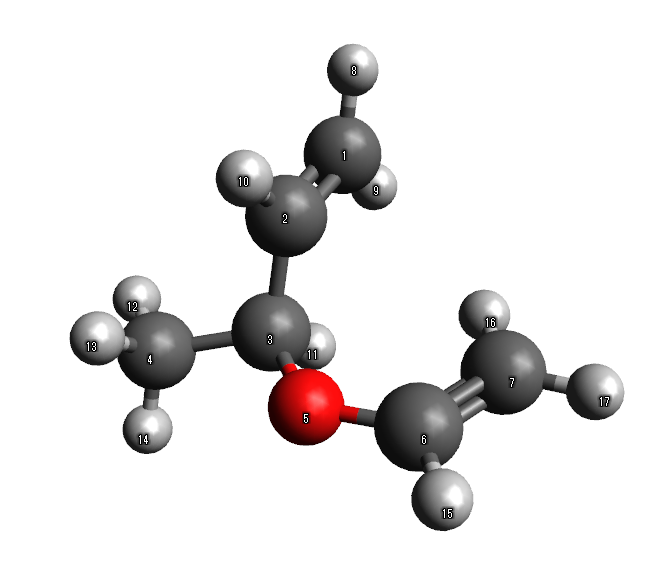
この操作を繰り返して、まずは結合次数が変わるものを入力します。

つづいて結合の生成消滅に対応します。まず、結合の生成です。結合ができる片側の原子からドラッグすると、新しい炭素原子が生まれ、ドラッグを始めた原子と結合を持った状態になります。

このまま結合を作りたい原子までドラッグすると、新しく生まれていた炭素原子が消え、ドラッグ元の原子とドラッグ先の原子間に単結合ができた状態となります。
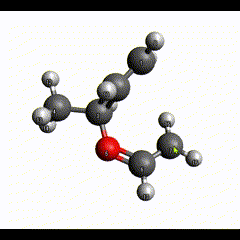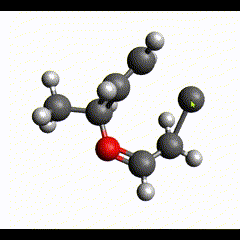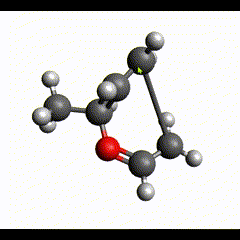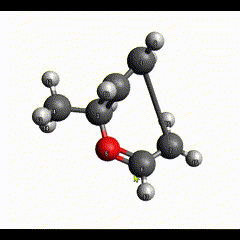
続いて結合を削除します。削除したい結合上で右クリックします。

これで結合情報を更新できました。残念ながらXYZファイルは結合情報はなく、このまま保存すると reactant.xyz と同じ分子にしかなりません。構造最適化してやります。
構造最適化ツールを選択、構造最適化してやります。

生成物の分子として妥当な構造になりました。これを File → Save As... を選択、 product.xyz として保存します。
一応、反応物 reactant.xyz と原子のラベルを突き合わせて、変な入れ替わりとかが起こってないかをチェックします。
こうやってできた reactant.xyz と product.xyz を、 CP2Kが使えるLinuxマシンに転送します。
CP2K入力ファイル
CP2Kがあれば計算できるといっても、その入力ファイルを作るのは一苦労です。そこでいろいろなチュートリアルやサンプルからコピーしてきます。
NEB計算は、反応物と生成物を結ぶビーズを並べ、それをばねでつないだような系を考えています。そしてその各ビーズごとのエネルギーを量子化学計算などで計算しています。CP2KではざっくりNEBのパートと量子化学計算のパートに分けられます。
プロジェクト設定も合わせると3つのパートに分けて、以下に書いてみます。
タイトル
これは CP2K のソースコードにあるテストファイルのうちNEBのテスト、cp2k/tests/NEB/regtest-1 フォルダ中の 2gly_IT-NEB.inp など、NEBのサンプルからとってきて、プロジェクト名を書き換えてやればよいです。
&GLOBAL
PRINT_LEVEL LOW
PROJECT claisen_rearrangement
RUN_TYPE BAND
&END GLOBAL
NEB計算
これも 2gly_IT-NEB.inp からとってきます。ただこれはビーズのエネルギーをMMで計算しているようなので、NEBに関する設定のみ取ってきます。
&MOTION
&BAND
BAND_TYPE IT-NEB
K_SPRING 0.05
NPROC_REP 1
NUMBER_OF_REPLICA 15
ROTATE_FRAMES T
&CONVERGENCE_CONTROL
#MAX_DR 0.01
MAX_FORCE 0.001
#RMS_DR 0.02
RMS_FORCE 0.0005
&END CONVERGENCE_CONTROL
&CONVERGENCE_INFO
&END CONVERGENCE_INFO
&OPTIMIZE_BAND
OPT_TYPE MD
&MD
MAX_STEPS 10
TEMPERATURE 100.0
TIMESTEP 0.5
&VEL_CONTROL
ANNEALING 0.95
PROJ_VELOCITY_VERLET T
&END VEL_CONTROL
#&TEMP_CONTROL
# TEMPeRATURE 100.0
# TEMP_TOL 50.
# TEMP_TOL_STEPS 100
#&END
&END MD
&END OPTIMIZE_BAND
&PROGRAM_RUN_INFO
&END PROGRAM_RUN_INFO
&REPLICA
COORD_FILE_NAME reactant.xyz
&END REPLICA
&REPLICA
COORD_FILE_NAME product.xyz
&END REPLICA
&END BAND
&PRINT
&VELOCITIES OFF
&END VELOCITIES
&END PRINT
&END MOTION
ざっくりと、15個のビーズを作り、MDのサンプリングを活用した最適化をしています。
生成物、反応物のビーズは2つの REPLICA で並べて指定しています。ここは 2gly-IT-NEB.inp から書き換えたところです。
何番目のビーズかを特に指定していないのですが、2つ指定した場合は始点と終点のビーズになるのかな?3つ以上指定すると指定した順番にビーズに振り分けるのでしょうか?ちゃんとドキュメントみなきゃ。
QM
今回の計算ではPM6とかがいいかなと思うのですが、残念ながらよさげなサンプルが見当たりませんでした。なので今回はCP2Kのドキュメントをにらんで自分で書きました。皆さんはぜひこれをたたき台にして各々の便利な入力テンプレートを作っていくとよいと思います。
&FORCE_EVAL
&DFT
&QS
METHOD PM6
&END QS
&SCF
# MAX_SCF 20
IGNORE_CONVERGENCE_FAILURE
SCF_GUESS ATOMIC
&END SCF
&END DFT
&SUBSYS
&CELL
ABC 30.0 30.0 30.0
PERIODIC NONE
&END CELL
&TOPOLOGY
COORDINATE XYZ
COORD_FILE_NAME reactant.xyz
&END TOPOLOGY
&END SUBSYS
&END FORCE_EVAL
IGNORE_CONVERGENCE_FAILURE は悩ましいのですが、NEBの最適化の特に初期段階では電子状態計算のSCFの収束に時間がかかることがあるので条件を緩めることにしました。
NEBの初期配置ビーズは反応物・生成物の各原子を直線で結んで等分していて、よくとんでもない原子間距離とかになったりします。そうすると場合によってはSCFが振動するとかもあり得えますので、収束ステップを大きくしたところでそのステップ数までふらふらするだけになりそうです。なので閾値とかの条件を緩めるのがよいかと思われます。まじめにやるならこの計算の結果を各ビーズの初期座標とし、量子化学計算の設定をもう少しきちっとやった高精度計算を行うのがよいでしょう。
CELL は周期境界条件の設定ですね。今回は真空中の1分子をイメージしているので PERIODIC NONE としました。 こうすると ABC とかセル長さは特に関係ないだろうと思って、分子を内部にもてるくらい十分大きな値を指定すればいいかなと 30 を指定しました。これもちゃんと確認しなきゃ。
TOPOLOGY では分子構造を指定しています。MOTION で既に product.xyz reactant.xyz を指定しているのにどうしてまた reatant.xyz が要るのだという気持ちになりますが、これはDFT計算にあたり必要な基底関数とか原子の数など、計算リソースの決定のために分子データを渡している感じです。実際、ほかの電子状態計算の例では、TOPOLOGY で基底関数に関する設定も行っていました。
NEB計算は各ビーズの座標を設定するのに REPRICA でファイルを指定しましたが、ここでは電子状態計算の設定のために渡している感じですね。なので結合情報とかは関係なく、元素の種類と数と順番が一致した XYZ ファイルであればなんでもよさそうです。
ファイル構成
とりあえず入力ファイル内容ができました。この3つの GLOBAL MOTION FORCE_EVAL を記録したファイルを claisen.inp とします。また同じディレクトリにAvogadroで作った reactant.xyz と product.xyz を置きます。
計算実行
試行1
とりあえずシングルプロセスでOpenMPで実行するバージョンでやってみます。
cp2k.ssmp claisen.inp > claisen.log
10ステップで収束し、AMD Ryzen 5 3600 6-Core で7秒程度かかりました。では結果を見てみます。まずエネルギープロファイルから。次のPythonプログラムをコマンドラインから実行してみます。
python -c "import matplotlib.pyplot as plt; import numpy as np; data = np.array([float(x) for x in open('claisen-rearrangement-1.ener').readlines()[-1].split()[1:] if x.startswith('-')])
plt.plot(range(1, len(data) + 1), (data - min(data))*2625.5, marker='o'); plt.xlabel('Bead index'); plt.ylabel('Energy/kJ/mol'); plt.grid(True); plt.savefig('graph.png')"
これで次のようなグラフができました。

活性化エネルギーが300 kJ/mol。ちょっと高すぎやしないか?と。
だいたいこれくらいの反応のNEBが10ステップだけというのはちょっと短すぎます。ということで計算閾値を工夫します。
試行2
ということで試行錯誤の結果次のようにしてみます。
MOTION/BAND セクション中にあるBANDの最適化に関する設定を以下のように変えます。
&OPTIMIZE_BAND
OPT_TYPE MD
&MD
MAX_STEPS 200
(以下略)
&END MD
&END OPTIMIZE_BAND
これでもう一度計算投入。ログファイルを見ると、今度は200ステップかかりました。単純にBANDの最適化で閾値まで収束せず、 MAX_STEP までかかっていますね。これでだいたい117秒、2分弱でした。
エネルギープロファイルはこのようになりました。

活性化エネルギーは125 kJ/mol くらい。まあこれくらいなら、計算の精度を考えたらあり得そう?
動画確認
続いて、反応機構を見ようと思います。CP2Kは各ビーズの最適化中のXYZ座標を各ファイルに出しますが、全ビーズのXYZ座標を順番に並べた便利なファイルは作っていません。これを次のコマンドで作ります。
for file in $(ls claisen_orig-pos-Replica_nr_*-1.xyz); do tail -n $(wc -l < reactant.xyz) $file >> traj.xyz; done
この traj.xyz をAvogadroで開いてみます。

まずは反応物の構造です。Avogadroの上部のメニューから Extensions → Animation... を選択します。

すると小さいウィンドウが出てきます。

ここで Dynamic Bonds にチェックを入れ、再生ボタンをクリックします。
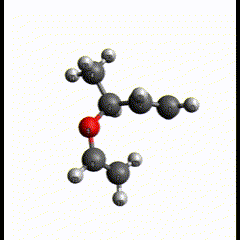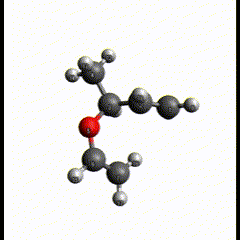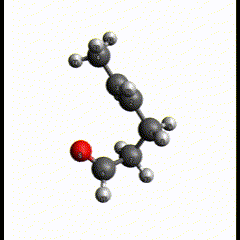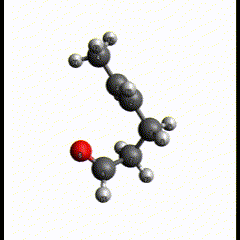
こんなふうに動いています。反応の切断と生成がほぼ同時っぽいので、まあ妥当な結果にはなっているかなと思います。
とくにエネルギーが高かったビーズは以下のような感じです:
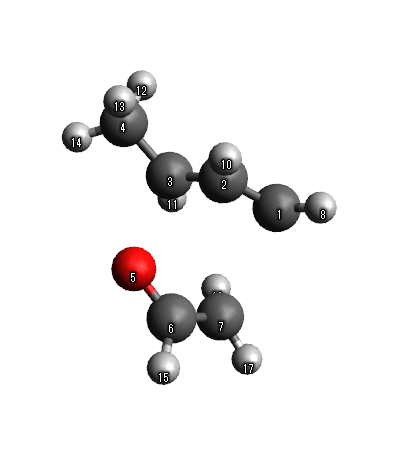
Wikipediaにあるような舟形かな?
これくらいの精度なら、これからどういう計算・実験をすればより詳しいことが分かりそうかというたたき台にはなるのではないでしょうか。
まとめ
化学反応の計算において強力な手段であるNEB計算を、(環境さえ整えれば)個人的に一番安く、手っ取り早く実行できると思っている手順を紹介しました。
まじめにこの記事において、CP2Kはともかく、Avogadroの構造作成方法は結構使えるかと思っています。これで反応物・生成物のXYZファイルさえ作れれば、CP2Kの他にもNWChemやGAMESSなども視野に入るかなと思います。
こんな感じで構造最適化をリアルタイムでやりつつマウスドラッグで分子を3Dで編集できるフリー or オープンなソフトウェアないかな・・・Avogadroの2はもうこの機能なさそうだし、あとはJMolを頑張るくらいしか・・・
皆さんの参考になれば幸いです。
Discussion