Myloasm: ナノポアリードとHiFiリードの両方に対応したde novoメタゲノムアセンブラー
はじめに

https://myloasm-docs.github.io/results/
Myloasmは、ロングリードシーケンスデータ用のde novoメタゲノムアセンブラです。
myloasmは以下のデータを対象としています。
- Nanopore R10 sinplex read
- 読み取り精度 >= ~97% (sup or hacでのベースコール)
- PacBio HiFiリード
開発背景として、メタゲノムは多種多様な生物のゲノムが混在しています。Long readのデータであっても、近縁株や変異株が混ざった環境下では、枝分かれ (bubble)/繰り返し/近縁株の混同がアセンブリの妨げになることがあります。
myloasmはこのようなメタゲノム特有の近縁・多変異株の混在する状況をうまく扱えるように設計されたアセンブラーです。

myloasmは以下のような原理に基づいて設計されています。
-
SNPを含むk-mer(SNPmer)の検出
- 通常のアセンブラーは、k-mer(長さkの連続塩基)を単純にノードとして扱いますが、myloasm は SNP(単一塩基多型)を含むk-mer を明示的に検出し、同一リード内での多様性情報を抽出します。これにより、株間変異を識別できる特徴量を初期段階から保持します。
-
SNP情報を組み込んだ文字列グラフの構築
- リード間のオーバーラップをノードとエッジで表現したstring graph(文字列グラフ)を構築します。SNPmer 情報を付与することで、近縁株由来のリードが異なる枝(分岐)として表現されます。これにより、通常のアセンブラーでは混同されるような株間の違いを構造的に保持します。
-
カバレッジと一致率に基づくエッジの重み付け
- 構築されたグラフ内で、各経路(パス)のリードカバレッジと配列同一性を算出し、一貫したカバレッジを持つ経路を高信頼経路として残す一方、低カバレッジや不均一な経路をノイズとして除去します。これにより、主要な株を表す安定した経路のみが保持されます。
-
株レベルでのアセンブリ生成
- 最終的に、整理されたグラフから株ごとに対応する経路を抽出し、重複の少ない高品質なコンティグを出力します。誤り訂正を過剰に行わないため、株間の多様性を失わずに再構築することが可能です。
詳細はプレプリントを参照下さい。
High-resolution metagenome assembly for modern long reads with myloasm
デモデータの取得
PacBioのデータはFTPサイトで公開されているもの、Nanoporeのデータはプレプリントで使用されていたものを使用します。
mkdir 00Fastq
# PacBio HiFi read
aria2c -x 16 -ctrue https://downloads.pacbcloud.com/public/dataset/Sequel-IIe-202104/metagenomics/m64011_210225_094432.hifi_reads.fastq.tar.gz -d 00Fastq
tar -xvf 00Fastq/m64011_210225_094432.hifi_reads.fastq.tar.gz -C 00Fastq
pigz -p 32 00Fastq/m64011_210225_094432.hifi_reads.fastq
# ONT R10.4
aria2c -x 16 -ctrue ftp://ftp.sra.ebi.ac.uk/vol1/fastq/DRR582/DRR582179/DRR582179_1.fastq.gz -d 00Fastq
aria2c -x 16 -ctrue ftp://ftp.sra.ebi.ac.uk/vol1/fastq/DRR582/DRR582205/DRR582205_1.fastq.gz -d 00Fastq
インストール
myloasmと結果確認などに使用するツールもインストールします。
# Miniforge3の導入
wget "https://github.com/conda-forge/miniforge/releases/download/25.3.0-3/Miniforge3-25.3.0-3-Linux-x86_64.sh"
bash Miniforge3-25.3.0-3-Linux-x86_64.sh -b -p $PWD/miniforge3
# activate
. ${PWD}/miniforge3/etc/profile.d/conda.sh
. ${PWD}/miniforge3/etc/profile.d/mamba.sh
conda activate
mamba create -n myloasm_env -c bioconda myloasm quast seqkit bandage bbmap checkm2 python=3.12 r-base r-seqinr r-ggplot2 r-dplyr
mamba activate myloasm_env
# Version
myloasm --version
# myloasm 0.2.0
checkm2はデータベースを取得する必要があります。
checkm2 databaseコマンドで取得します。--downloadオプションに--pathオプションで特定の場所に配置できます。カレントディレクトリに配置するなら下記のように実行。
checkm2 database --download --path ./
[10/26/2025 09:42:51 PM] INFO: Command: Download database. Checking internal path information.
[10/26/2025 09:42:54 PM] INFO: Downloading https://zenodo.org/api/records/14897628/files/checkm2_database.tar.gz/content to ./checkm2_database.tar.gz.
100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1.74G/1.74G [16:56<00:00, 1.71MiB/s]
[10/26/2025 09:59:54 PM] INFO: Extracting files from archive...
[10/26/2025 10:00:09 PM] INFO: Verifying version and checksums...
[10/26/2025 10:00:13 PM] INFO: Verification success.
[10/26/2025 10:00:15 PM] INFO: Diamond DATABASE downloaded successfully! Consider running <checkm2 testrun> to verify everything works.
コマンドフレーズは以下のとおりです。
myloasm reads1.fq reads2.fq reads3.fq -o output_directory -t 32
ストレージ容量が多くない場合、--clean-dirを使用すると中間ファイルが生成されない?みたいです。また、精度は落ちますが、-cの値を調整することで、RAMの使用量を抑えることができるようです。
USAGE myloasm
myloasm - high-resolution metagenomic assembly with noisy long reads. See online documentation for full options.
EXAMPLE (Nanopore R10): myloasm nanopore_reads.fq.gz -o output_directory -t 50
EXAMPLE (PacBio HiFi): myloasm pacbio_reads.fq.gz -o output_directory -t 50 --hifi
Usage: myloasm [OPTIONS] <FASTQ/FASTA (.gz)>...
Arguments:
<FASTQ/FASTA (.gz)>... Input read file(s) -- multiple files are concatenated
Options:
-o, --output-dir <OUTPUT_DIR> Output directory for results; created if it does not exist [default: myloasm-out]
-t, --threads <THREADS> Number of threads to use for processing [default: 20]
--clean-dir Do not dump large intermediate data to disk (intermediate data is useful for rerunning)
-l, --log-level <LOG_LEVEL> Verbosity level. Warning: trace is very verbose [default: debug] [possible values: error, warn, info, debug, trace]
-h, --help Print help
-V, --version Print version
Technology Presets:
--nano-r10 (DEFAULT) R10 nanopore mode for sup/hac data (> ~97% median accuracy). Specifying this flag does not do anything for now
--hifi PacBio HiFi mode -- assumes less chimericism and higher accuracy
Basic Algorithmic Parameters:
-c, --c <C>
Compression ratio (1/c k-mers selected). Must be <= 15 [default: 11]
--quality-value-cutoff <QUALITY_VALUE_CUTOFF>
Disallow reads with < % identity for graph building (estimated from base qualities) [default: 90]
--min-ol <MIN_OL>
Minimum overlap length for graph construction [default: 500]
-b, --bloom-filter-size <BLOOM_FILTER_SIZE>
Bloom filter size in GB. Increase for massive datasets [default: 10]
--aggressive-bloom
More aggressive filtering of low-abundance k-mers. May be non-deterministic
Output thresholds:
--min-reads-contig <MIN_READS_CONTIG>
Output contigs with >= this number of reads [default: 1]
--singleton-coverage-threshold <SINGLETON_COVERAGE_THRESHOLD>
Remove singleton contigs with <= this estimated coverage depth (DP1 coverage; 99% identity coverage) [default: 3]
--secondary-coverage-threshold <SECONDARY_COVERAGE_THRESHOLD>
Remove contigs with <= this estimated coverage depth and <= 2 reads (DP1 coverage; 99% identity coverage) [default: 1]
--absolute-coverage-threshold <ABSOLUTE_COVERAGE_THRESHOLD>
Remove all contigs with <= this estimated coverage depth (DP1 coverage; 99% identity coverage)
myloasmの実行
Nanoporeリード
複数のデータを1コマンドで実行してみます。
myloasm 00Fastq/DRR582179_1.fastq.gz 00Fastq/DRR582205_1.fastq.gz -o myloasm_results/nanopore -t 32 --log-level info --nano-r10
出力
(2025-10-26 01:06:08.648) INFO [myloasm] COMMAND: myloasm 00Fastq/DRR582179_1.fastq.gz 00Fastq/DRR582205_1.fastq.gz -o myloasm_results/nanopore -t 32 --log-level info --nano-r10
(2025-10-26 01:06:08.649) INFO [myloasm] VERSION: 0.2.0
(2025-10-26 01:06:08.649) INFO [myloasm] SYSTEM NAME: Ubuntu
(2025-10-26 01:06:08.649) INFO [myloasm] SYSTEM HOST NAME: <UserName>
(2025-10-26 01:06:08.651) INFO [myloasm] Starting assembly...
thread '<unnamed>' panicked at src/seq_parse.rs:100:38:
Error reading record: ParseError { msg: "Sequence length is 2296 but quality length is 2497", kind: UnequalLengths, position: ErrorPosition { line: 159581, id: Some("DRR582179.39896") }, format: Some(Fastq) }
note: run with RUST_BACKTRACE=1 environment variable to display a backtrace
(2025-10-26 01:06:32.801) INFO [myloasm::seq_parse] Finished with bloom filter processing in 24.149586901s. Round 2 - Start k-mer counting...
(2025-10-26 01:06:32.864) INFO [myloasm::utils] Memory usage after bloom filter processing --- Memory usage: 1.56 GB
thread '<unnamed>' panicked at src/seq_parse.rs:254:34:
Error reading record: ParseError { msg: "Sequence length is 2296 but quality length is 2497", kind: UnequalLengths, position: ErrorPosition { line: 159581, id: Some("DRR582179.39896") }, format: Some(Fastq) }
(2025-10-26 01:06:37.786) INFO [myloasm::seq_parse] Finished with k-mer counting in 4.92206169s. Total kmers after bloom filter: 53994783
(2025-10-26 01:06:37.844) INFO [myloasm::utils] Memory usage after second round of k-mer counting processing --- Memory usage: 1.56 GB
(2025-10-26 01:06:38.833) INFO [myloasm::seq_parse] Removed 15495151 kmers with counts < 1 in both strands and <= 3 multiplicity.
(2025-10-26 01:06:38.878) INFO [myloasm] Time elapsed in for counting k-mers is: 30.226542005s
(2025-10-26 01:06:39.172) INFO [myloasm::kmer_comp] Finding snpmers...
(2025-10-26 01:06:40.258) INFO [myloasm] Time elapsed in for parsing snpmers is: 1.379672884s
(2025-10-26 01:06:40.302) INFO [myloasm::utils] STAGE 1: Obtained SNPmers --- Memory usage: 0.04 GB
(2025-10-26 01:06:40.303) INFO [myloasm] Getting twin reads from snpmers...
thread '<unnamed>' panicked at src/kmer_comp.rs:68:34:
Error reading record: ParseError { msg: "Sequence length is 2296 but quality length is 2497", kind: UnequalLengths, position: ErrorPosition { line: 159581, id: Some("DRR582179.39896") }, format: Some(Fastq) }
thread '<unnamed>' panicked at src/kmer_comp.rs:68:34:
Error reading record: ParseError { msg: "Expected '+' separator but found 'G", kind: InvalidSeparator, position: ErrorPosition { line: 750563, id: Some("DRR582205.187641") }, format: Some(Fastq) }
(2025-10-26 01:06:51.947) INFO [myloasm::kmer_comp] Number of valid reads with >= 1kb - 210442. Number of reads below quality threshold - 0.
(2025-10-26 01:06:51.949) INFO [myloasm::kmer_comp] Mean SNPmer density: 0.41%
(2025-10-26 01:06:51.949) INFO [myloasm::kmer_comp] Time elapsed for obtaining twin reads is: 11.646420106s
(2025-10-26 01:06:52.028) INFO [myloasm::utils] STAGE 1.5: Initially obtained dirty twin reads --- Memory usage: 2.28 GB
(2025-10-26 01:06:52.028) INFO [myloasm] Removing contained reads - round 1...
(2025-10-26 01:07:30.198) INFO [myloasm::twin_graph] 152534 reads are contained; 57908 outer reads
(2025-10-26 01:07:30.198) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 38.169735255s
(2025-10-26 01:07:30.603) INFO [myloasm] Mapping reads to non-contained (outer) reads - round 1...
(2025-10-26 01:07:30.603) INFO [myloasm::mapping] Mapping 210442 reads to 57908 outer reads
(2025-10-26 01:08:27.863) INFO [myloasm::mapping] Number of local alignments to outer reads: 6489541
(2025-10-26 01:08:27.863) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 1141581
(2025-10-26 01:08:27.979) INFO [myloasm::utils] STAGE 2: Finished mapping reads to outer reads --- Memory usage: 4.09 GB
(2025-10-26 01:08:27.980) INFO [myloasm] Processing mappings to clean and split chimeric reads...
(2025-10-26 01:08:34.098) INFO [myloasm] Initial chimera detection: split 28480 possibly chimeric or noisy reads out of 218894 total reads (13.01%)
(2025-10-26 01:08:34.098) INFO [myloasm] Removing contained reads after splitting ...
(2025-10-26 01:09:11.472) INFO [myloasm::twin_graph] 171435 reads are contained; 47459 outer reads
(2025-10-26 01:09:11.472) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 37.373674094s
(2025-10-26 01:09:11.863) INFO [myloasm] Round 2: Mapping reads to 10853 candidate outer reads...
(2025-10-26 01:09:11.863) INFO [myloasm::mapping] Mapping 218894 reads to 10853 outer reads
(2025-10-26 01:09:38.708) INFO [myloasm::mapping] Number of local alignments to outer reads: 2202615
(2025-10-26 01:09:38.708) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 457763
(2025-10-26 01:09:38.708) INFO [myloasm] Round 2: Processing mappings...
(2025-10-26 01:09:40.096) INFO [myloasm] Round 2: Gained 640 reads after splitting chimeras and mapping to outer reads in 28.238562691s
(2025-10-26 01:09:40.096) INFO [myloasm] Round 2: Removing contained reads after splitting ...
(2025-10-26 01:09:57.408) INFO [myloasm::twin_graph] 4187 reads are contained; 45175 outer reads
(2025-10-26 01:09:57.408) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 17.311963186s
(2025-10-26 01:09:57.712) INFO [myloasm] Final round: Mapping reads to 10853 candidate outer reads...
(2025-10-26 01:09:57.712) INFO [myloasm::mapping] Mapping 219534 reads to 1092 outer reads
(2025-10-26 01:10:05.614) INFO [myloasm::mapping] Number of local alignments to outer reads: 240587
(2025-10-26 01:10:05.614) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 34349
(2025-10-26 01:10:07.215) INFO [myloasm::utils] STAGE 3: Obtained clean twin reads --- Memory usage: 2.71 GB
(2025-10-26 01:10:07.215) INFO [myloasm] Getting overlaps between outer reads...
(2025-10-26 01:10:24.892) INFO [myloasm] Time elapsed for getting overlaps is: 17.677827393s
(2025-10-26 01:10:25.246) INFO [myloasm] Initial light cleaning...
(2025-10-26 01:10:27.438) INFO [myloasm] Heavy graph cleaning...
(2025-10-26 01:10:37.575) INFO [myloasm::small_genomes] Small circular contig extraction...
(2025-10-26 01:10:43.019) INFO [myloasm] Progressive coverage filtering...
(2025-10-26 01:10:47.831) INFO [myloasm] Dereplicating spurious contigs...
(2025-10-26 01:11:19.964) INFO [myloasm::utils] STAGE 4: Obtained unpolished contigs --- Memory usage: 4.06 GB
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] Unpolished assembly statistics - ROUGH ESTIMATE; NOT FINAL
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) N50: 173727
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Largest contig has size: 2492792
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Number of contigs: 2258
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Number of possibly circular contigs >= 1M: 9
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Number of contigs (circular or non-circular)>= 1M: 28
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Number of contigs >= 100k: 408
(2025-10-26 01:11:19.964) INFO [myloasm::unitig] (Estimated) Total bases within assembly is 194526004
(2025-10-26 01:11:19.974) INFO [myloasm] Beginning final alignment of reads to graph...
(2025-10-26 01:11:31.456) INFO [myloasm::mapping] Index built; starting mapping
(2025-10-26 01:11:33.461) INFO [myloasm::mapping] Mapped 10% of reads back to contigs
(2025-10-26 01:11:35.690) INFO [myloasm::mapping] Mapped 20% of reads back to contigs
(2025-10-26 01:11:38.024) INFO [myloasm::mapping] Mapped 30% of reads back to contigs
(2025-10-26 01:11:39.926) INFO [myloasm::mapping] Mapped 40% of reads back to contigs
(2025-10-26 01:11:42.063) INFO [myloasm::mapping] Mapped 50% of reads back to contigs
(2025-10-26 01:11:44.439) INFO [myloasm::mapping] Mapped 60% of reads back to contigs
(2025-10-26 01:11:46.475) INFO [myloasm::mapping] Mapped 70% of reads back to contigs
(2025-10-26 01:11:48.640) INFO [myloasm::mapping] Mapped 80% of reads back to contigs
(2025-10-26 01:11:50.957) INFO [myloasm::mapping] Mapped 90% of reads back to contigs
(2025-10-26 01:11:53.985) INFO [myloasm::mapping] Mapped 100% of reads back to contigs
(2025-10-26 01:11:54.525) INFO [myloasm::utils] STAGE 5: Mapped reads to contigs --- Memory usage: 3.54 GB
(2025-10-26 01:11:54.525) INFO [myloasm] Time elapsed for aligning reads to graph is 34.551590169s
(2025-10-26 01:11:54.526) INFO [myloasm] Polishing final contigs...
(2025-10-26 01:12:58.769) INFO [myloasm::polishing_mod] Polished 10% of contigs...
(2025-10-26 01:13:31.431) INFO [myloasm::polishing_mod] Polished 20% of contigs...
(2025-10-26 01:13:47.912) INFO [myloasm::polishing_mod] Polished 30% of contigs...
(2025-10-26 01:14:02.765) INFO [myloasm::polishing_mod] Polished 40% of contigs...
(2025-10-26 01:14:32.444) INFO [myloasm::polishing_mod] Polished 50% of contigs...
(2025-10-26 01:15:24.544) INFO [myloasm::polishing_mod] Polished 60% of contigs...
(2025-10-26 01:16:12.478) INFO [myloasm::polishing_mod] Polished 70% of contigs...
(2025-10-26 01:16:31.008) INFO [myloasm::polishing_mod] Polished 80% of contigs...
(2025-10-26 01:16:53.473) INFO [myloasm::polishing_mod] Polished 90% of contigs...
(2025-10-26 01:18:52.570) INFO [myloasm] Time elapsed for polishing is 452.595679652s
(2025-10-26 01:18:52.570) INFO [myloasm] Dereplicating polished contigs with skani...
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] -------------- FINAL primary assembly statistics --------------
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] N50: 191655
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Largest contig has size: 2453227
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Number of primary contigs: 2048
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Number of alt/dup contigs: 122 and 72
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Number of possibly circular contigs >= 1M: 9
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Number of contigs >= 1M: 28
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Number of contigs >= 100k: 407
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] Total bases within assembly is 188341585
(2025-10-26 01:18:56.640) INFO [myloasm::skani_dereplicate] -------------------------------------------------
(2025-10-26 01:18:56.679) INFO [myloasm] Assembly completed. Total time elapsed is 768.030899431s
一部のリードにエラーがあったようですが、ツールの問題ではなさそうなので今回は無視します。
PacBio HiFiリード
myloasm 00Fastq/m64011_210225_094432.hifi_reads.fastq.gz -o myloasm_results/pac -t 32 --hifi
出力
(2025-10-25 20:09:46.340) INFO [myloasm] COMMAND: myloasm 00Fastq/m64011_210225_094432.hifi_reads.fastq.gz -o myloasm_results/pac -t 32 --hifi
(2025-10-25 20:09:46.340) INFO [myloasm] VERSION: 0.2.0
(2025-10-25 20:09:46.340) INFO [myloasm] SYSTEM NAME: Ubuntu
(2025-10-25 20:09:46.340) INFO [myloasm] SYSTEM HOST NAME: <UserName>
(2025-10-25 20:09:46.343) INFO [myloasm] Starting assembly...
(2025-10-25 20:10:35.120) INFO [myloasm::seq_parse] Processed 100000 reads.
(2025-10-25 20:11:33.422) INFO [myloasm::seq_parse] Processed 200000 reads.
(2025-10-25 20:12:34.211) INFO [myloasm::seq_parse] Processed 300000 reads.
(2025-10-25 20:13:37.838) INFO [myloasm::seq_parse] Processed 400000 reads.
(2025-10-25 20:14:42.232) INFO [myloasm::seq_parse] Processed 500000 reads.
(2025-10-25 20:15:46.236) INFO [myloasm::seq_parse] Processed 600000 reads.
(2025-10-25 20:16:51.213) INFO [myloasm::seq_parse] Processed 700000 reads.
(2025-10-25 20:17:56.930) INFO [myloasm::seq_parse] Processed 800000 reads.
(2025-10-25 20:19:02.232) INFO [myloasm::seq_parse] Processed 900000 reads.
(2025-10-25 20:20:06.027) INFO [myloasm::seq_parse] Processed 1000000 reads.
(2025-10-25 20:21:10.844) INFO [myloasm::seq_parse] Processed 1100000 reads.
(2025-10-25 20:22:18.248) INFO [myloasm::seq_parse] Processed 1200000 reads.
(2025-10-25 20:23:24.741) INFO [myloasm::seq_parse] Processed 1300000 reads.
(2025-10-25 20:24:30.907) INFO [myloasm::seq_parse] Processed 1400000 reads.
(2025-10-25 20:25:37.238) INFO [myloasm::seq_parse] Processed 1500000 reads.
(2025-10-25 20:26:42.827) INFO [myloasm::seq_parse] Processed 1600000 reads.
(2025-10-25 20:27:40.109) INFO [myloasm::seq_parse] Finished with bloom filter processing in 1073.765921991s. Round 2 - Start k-mer counting...
(2025-10-25 20:27:40.306) INFO [myloasm::utils] Memory usage after bloom filter processing --- Memory usage: 19.45 GB
(2025-10-25 20:31:11.793) INFO [myloasm::seq_parse] Finished with k-mer counting in 211.486657345s. Total kmers after bloom filter: 701807901
(2025-10-25 20:31:11.996) INFO [myloasm::utils] Memory usage after second round of k-mer counting processing --- Memory usage: 19.45 GB
(2025-10-25 20:31:27.688) INFO [myloasm::seq_parse] Removed 84478090 kmers with counts < 1 in both strands and <= 3 multiplicity.
(2025-10-25 20:31:27.910) INFO [myloasm] Time elapsed in for counting k-mers is: 1301.566788604s
(2025-10-25 20:31:32.399) INFO [myloasm::kmer_comp] Finding snpmers...
(2025-10-25 20:31:57.082) INFO [myloasm] Time elapsed in for parsing snpmers is: 29.172522413s
(2025-10-25 20:31:57.408) INFO [myloasm::utils] STAGE 1: Obtained SNPmers --- Memory usage: 0.40 GB
(2025-10-25 20:31:57.408) INFO [myloasm] Getting twin reads from snpmers...
(2025-10-25 20:33:08.489) INFO [myloasm::kmer_comp] Number of valid reads with >= 1kb - 1687208. Number of reads below quality threshold - 0.
(2025-10-25 20:33:08.508) INFO [myloasm::kmer_comp] Mean SNPmer density: 2.33%
(2025-10-25 20:33:08.508) INFO [myloasm::kmer_comp] Time elapsed for obtaining twin reads is: 71.099874918s
(2025-10-25 20:33:08.869) INFO [myloasm::utils] STAGE 1.5: Initially obtained dirty twin reads --- Memory usage: 18.51 GB
(2025-10-25 20:33:08.869) INFO [myloasm] Removing contained reads - round 1...
(2025-10-25 20:36:18.747) INFO [myloasm::twin_graph] 1310057 reads are contained; 377151 outer reads
(2025-10-25 20:36:18.747) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 189.87843088s
(2025-10-25 20:36:21.053) INFO [myloasm] Mapping reads to non-contained (outer) reads - round 1...
(2025-10-25 20:36:21.053) INFO [myloasm::mapping] Mapping 1687208 reads to 377151 outer reads
(2025-10-25 20:45:50.821) INFO [myloasm::mapping] Number of local alignments to outer reads: 128656359
(2025-10-25 20:45:50.821) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 23707347
(2025-10-25 20:45:51.290) INFO [myloasm::utils] STAGE 2: Finished mapping reads to outer reads --- Memory usage: 22.99 GB
(2025-10-25 20:45:51.290) INFO [myloasm] Processing mappings to clean and split chimeric reads...
(2025-10-25 20:46:52.430) INFO [myloasm] Initial chimera detection: split 68001 possibly chimeric or noisy reads out of 1717285 total reads (3.96%)
(2025-10-25 20:46:52.430) INFO [myloasm] Removing contained reads after splitting ...
(2025-10-25 20:50:04.341) INFO [myloasm::twin_graph] 1380192 reads are contained; 337093 outer reads
(2025-10-25 20:50:04.341) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 191.91113505s
(2025-10-25 20:50:06.806) INFO [myloasm] Round 2: Mapping reads to 30009 candidate outer reads...
(2025-10-25 20:50:06.806) INFO [myloasm::mapping] Mapping 1717285 reads to 30009 outer reads
(2025-10-25 20:50:36.480) INFO [myloasm::mapping] Number of local alignments to outer reads: 9454388
(2025-10-25 20:50:36.480) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 1290796
(2025-10-25 20:50:36.480) INFO [myloasm] Round 2: Processing mappings...
(2025-10-25 20:50:40.809) INFO [myloasm] Round 2: Gained 1745 reads after splitting chimeras and mapping to outer reads in 34.058233486s
(2025-10-25 20:50:40.809) INFO [myloasm] Round 2: Removing contained reads after splitting ...
(2025-10-25 20:51:26.827) INFO [myloasm::twin_graph] 43322 reads are contained; 327659 outer reads
(2025-10-25 20:51:26.827) INFO [myloasm::twin_graph] Time elapsed for removing contained reads is: 46.010140275s
(2025-10-25 20:51:28.105) INFO [myloasm] Final round: Mapping reads to 30009 candidate outer reads...
(2025-10-25 20:51:28.105) INFO [myloasm::mapping] Mapping 1719030 reads to 2720 outer reads
(2025-10-25 20:51:32.478) INFO [myloasm::mapping] Number of local alignments to outer reads: 578426
(2025-10-25 20:51:32.478) INFO [myloasm::mapping] Number of maximal alignments to outer reads: 79937
(2025-10-25 20:51:46.129) INFO [myloasm::utils] STAGE 3: Obtained clean twin reads --- Memory usage: 18.05 GB
(2025-10-25 20:51:46.129) INFO [myloasm] Getting overlaps between outer reads...
(2025-10-25 20:54:49.541) INFO [myloasm] Time elapsed for getting overlaps is: 183.412817664s
(2025-10-25 20:55:00.613) INFO [myloasm] Initial light cleaning... (2025-10-25 20:58:04.913) INFO [myloasm::small_genomes] Small circular contig extraction...
(2025-10-25 20:58:26.987) INFO [myloasm] Progressive coverage filtering...
(2025-10-25 20:59:12.387) INFO [myloasm] Dereplicating spurious contigs...
(2025-10-25 21:02:51.008) INFO [myloasm::utils] STAGE 4: Obtained unpolished contigs --- Memory usage: 19.29 GB
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] Unpolished assembly statistics - ROUGH ESTIMATE; NOT FINAL
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) N50: 177768
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Largest contig has size: 6348821
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Number of contigs: 17888
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Number of possibly circular contigs >= 1M: 48
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Number of contigs (circular or non-circular)>= 1M: 168
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Number of contigs >= 100k: 1545
(2025-10-25 21:02:51.011) INFO [myloasm::unitig] (Estimated) Total bases within assembly is 1253379240
(2025-10-25 21:02:51.202) INFO [myloasm] Beginning final alignment of reads to graph...
(20(2025-10-25 21:06:29.358) INFO [myloasm::mapping] Mapped 10% of reads back to contigs
(2025-10-25 21:08:14.125) INFO [myloasm::mapping] Mapped 20% of reads back to contigs
(2025-10-25 21:09:59.372) INFO [myloasm::mapping] Mapped 30% of reads back to contigs
(2025-10-25 21:11:43.595) INFO [myloasm::mapping] Mapped 40% of reads back to contigs
(2025-10-25 21:13:26.940) INFO [myloasm::mapping] Mapped 50% of reads back to contigs
(2025-10-25 21:15:10.687) INFO [myloasm::mapping] Mapped 60% of reads back to contigs
(2025-10-25 21:16:54.063) INFO [myloasm::mapping] Mapped 70% of reads back to contigs
(2025-10-25 21:18:37.387) INFO [myloasm::mapping] Mapped 80% of reads back to contigs
(2025-10-25 21:20:21.585) INFO [myloasm::mapping] Mapped 90% of reads back to contigs
(2025-10-25 21:22:05.831) INFO [myloasm::mapping] Mapped 100% of reads back to contigs
(2025-10-25 21:22:11.682) INFO [myloasm::utils] STAGE 5: Mapped reads to contigs --- Memory usage: 23.18 GB
(2025-10-25 21:22:11.682) INFO [myloasm] Time elapsed for aligning reads to graph is 1160.479306658s
(2025-10-25 21:22:11.682) INFO [myloasm] Polishing final contigs...
(2025-10-25 21:39:45.049) INFO [myloasm::polishing_mod] Polished 10% of contigs...
(2025-10-25 21:42:33.804) INFO [myloasm::polishing_mod] Polished 20% of contigs...
(2025-10-25 21:44:20.907) INFO [myloasm::polishing_mod] Polished 30% of contigs...
(2025-10-25 21:45:39.152) INFO [myloasm::polishing_mod] Polished 40% of contigs...
(2025-10-25 21:46:45.314) INFO [myloasm::polishing_mod] Polished 50% of contigs...
(2025-10-25 21:47:44.072) INFO [myloasm::polishing_mod] Polished 60% of contigs...
(2025-10-25 21:48:34.164) INFO [myloasm::polishing_mod] Polished 70% of contigs...
(2025-10-25 21:49:19.120) INFO [myloasm::polishing_mod] Polished 80% of contigs...
(2025-10-25 21:50:01.085) INFO [myloasm::polishing_mod] Polished 90% of contigs...
(2025-10-25 21:59:41.382) INFO [myloasm::polishing_mod] Polished 100% of contigs...
(2025-10-25 21:59:44.376) INFO [myloasm] Time elapsed for polishing is 3413.173979346s
(2025-10-25 21:59:44.376) INFO [myloasm] Dereplicating polished contigs with skani...
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] -------------- FINAL primary assembly statistics --------------
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] N50: 252779
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Largest contig has size: 6341195
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Number of primary contigs: 14626
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Number of alt/dup contigs: 2573 and 683
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Number of possibly circular contigs >= 1M: 48
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Number of contigs >= 1M: 168
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Number of contigs >= 100k: 1540
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] Total bases within assembly is 1158132778
(2025-10-25 22:00:04.003) INFO [myloasm::skani_dereplicate] -------------------------------------------------
(2025-10-25 22:00:04.238) INFO [myloasm] Assembly completed. Total time elapsed is 6617.898315971s
PacBioのデータは特にエラーなく終了。ただ、Nanoporeのデータより多くの時間を要しました。
出力
myloasm_results下のNanoporeとPacBio用フォルダ下に結果ファイルが出力されます。
tree myloasm_results/ -L 2 -ha
[4.0K] myloasm_results/
├── [4.0K] nanopore
│ ├── [4.0K] 0-cleaning_and_unitigs
│ ├── [4.0K] 1-light_resolve
│ ├── [4.0K] 2-heavy_path_resolve
│ ├── [4.0K] 3-mapping
│ ├── [4.0K] alternate_assemblies
│ ├── [4.0K] assembly_graphs
│ ├── [180M] assembly_primary.fa
│ ├── [4.0K] binary_temp
│ ├── [9.9K] final_contig_graph.edges
│ ├── [1.9M] final_contig_graph.gfa
│ └── [9.2K] myloasm_2025-10-26_01-06-08.log
└── [4.0K] pac
├── [4.0K] 0-cleaning_and_unitigs
├── [4.0K] 1-light_resolve
├── [4.0K] 2-heavy_path_resolve
├── [4.0K] 3-mapping
├── [4.0K] alternate_assemblies
├── [4.0K] assembly_graphs
├── [1.1G] assembly_primary.fa
├── [4.0K] binary_temp
├── [ 86K] final_contig_graph.edges
├── [ 39M] final_contig_graph.gfa
└── [ 86K] myloasm_2025-10-25_20-09-46.log
16 directories, 8 files
Nanoporeは2サンプルのデータを一括で解析しましたが、結果ファイルは基本的に1つのようです。デフォルトでCoassemblyが実行されるようです。
コンティグファイル: assemble_primary.fa
後続解析で使用するコンティグfastaファイルです。seqkit statsとBBtoolsのstatsで結果確認します。
- BBtools
PacBioのデータ。
stats.sh in=myloasm_results/pac/assembly_primary.fa
A C G T N IUPAC Other GC GC_stdev
0.2644 0.2359 0.2343 0.2653 0.0000 0.0000 0.0000 0.4703 0.0880
Main genome scaffold total: 14626
Main genome contig total: 14626
Main genome scaffold sequence total: 1158.133 Mb
Main genome contig sequence total: 1158.133 Mb 0.000% gap
Main genome scaffold N/L50: 621/249.402 Kbp
Main genome contig N/L50: 621/249.402 Kbp
Main genome scaffold N/L90: 8570/27.434 Kbp
Main genome contig N/L90: 8570/27.434 Kbp
Max scaffold length: 6.341 Mbp
Max contig length: 6.341 Mbp
Number of scaffolds > 50 KB: 3863
% main genome in scaffolds > 50 KB: 75.22%
Minimum Number Number Total Total Scaffold
Scaffold of of Scaffold Contig Contig
Length Scaffolds Contigs Length Length Coverage
-------- -------------- -------------- -------------- -------------- --------
All 14,626 14,626 1,158,132,778 1,158,132,778 100.00%
1 Kbp 14,626 14,626 1,158,132,778 1,158,132,778 100.00%
2.5 Kbp 14,574 14,574 1,158,051,667 1,158,051,667 100.00%
5 Kbp 14,540 14,540 1,157,917,491 1,157,917,491 100.00%
10 Kbp 14,256 14,256 1,155,613,950 1,155,613,950 100.00%
25 Kbp 9,504 9,504 1,066,814,168 1,066,814,168 100.00%
50 Kbp 3,863 3,863 871,108,927 871,108,927 100.00%
100 Kbp 1,482 1,482 708,324,503 708,324,503 100.00%
250 Kbp 619 619 578,651,535 578,651,535 100.00%
500 Kbp 338 338 482,054,754 482,054,754 100.00%
1 Mbp 168 168 363,968,214 363,968,214 100.00%
2.5 Mbp 52 52 174,393,895 174,393,895 100.00%
5 Mbp 1 1 6,341,195 6,341,195 100.00%
Nanoporeのデータ。
stats.sh in=myloasm_results/nanopore/assembly_primary.fa
A C G T N IUPAC Other GC GC_stdev
0.2785 0.2217 0.2212 0.2785 0.0000 0.0000 0.0000 0.4430 0.0931
Main genome scaffold total: 2048
Main genome contig total: 2048
Main genome scaffold sequence total: 188.342 Mb
Main genome contig sequence total: 188.342 Mb 0.000% gap
Main genome scaffold N/L50: 160/191.655 Kbp
Main genome contig N/L50: 160/191.655 Kbp
Main genome scaffold N/L90: 1102/37.935 Kbp
Main genome contig N/L90: 1102/37.935 Kbp
Max scaffold length: 2.453 Mbp
Max contig length: 2.453 Mbp
Number of scaffolds > 50 KB: 880
% main genome in scaffolds > 50 KB: 84.82%
Minimum Number Number Total Total Scaffold
Scaffold of of Scaffold Contig Contig
Length Scaffolds Contigs Length Length Coverage
-------- -------------- -------------- -------------- -------------- --------
All 2,048 2,048 188,341,585 188,341,585 100.00%
1 Kbp 2,048 2,048 188,341,585 188,341,585 100.00%
2.5 Kbp 2,016 2,016 188,289,517 188,289,517 100.00%
5 Kbp 1,967 1,967 188,107,112 188,107,112 100.00%
10 Kbp 1,835 1,835 187,073,256 187,073,256 100.00%
25 Kbp 1,440 1,440 180,184,776 180,184,776 100.00%
50 Kbp 880 880 159,743,933 159,743,933 100.00%
100 Kbp 397 397 125,219,788 125,219,788 100.00%
250 Kbp 108 108 82,997,558 82,997,558 100.00%
500 Kbp 54 54 65,297,302 65,297,302 100.00%
1 Mbp 28 28 47,012,111 47,012,111 100.00%
seqkit stats -T myloasm_results/pac/assembly_primary.fa myloasm_results/nanopore/assembly_primary.fa
| file | format | type | num_seqs | sum_len | min_len | avg_len | max_len | Q1 | Q2 | Q3 | sum_gap | N50 | N50_num | Q20(%) | Q30(%) | AvgQual | GC(%) | sum_n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pac/assembly_primary.fa | FASTA | DNA | 14626 | 1158132778 | 1056 | 79183.2 | 6341195 | 21291 | 31259 | 51932 | 0 | 249402 | 620 | 0 | 0 | 0.00 | 47.03 | 0 |
| nanopore/assembly_primary.fa | FASTA | DNA | 2048 | 188341585 | 1035 | 91963.7 | 2453227 | 21164 | 42041 | 83888 | 0 | 191655 | 160 | 0 | 0 | 0.00 | 44.30 | 0 |
Pacのデータで過去に実施したアセンブリの結果です。
| Num seqs | min len | max len | ave len | N50 | |
|---|---|---|---|---|---|
| hifiasm_meta | 11,693 | 4,918 | 4,413,267 | 98,364.8 | 302,611 |
| metaflye | 10,419 | 518 | 4,412,735 | 96,945.0 | 181,422 |
| metaMDBG | 26,769 | 510 | 7,383,977 | 43,713.0 | 201,253 |
過去の結果と同様に分布の視覚化をして見たところ、hifiasm_metaと類似した分布を示していました。
plot script
# load package
require(seqinr)
require(ggplot2)
require(dplyr)
# import contig
myloasm_contig <- seqinr::read.fasta("myloasm_results/pac/assembly_primary.fa")
# Obtain contig length summary
myloasm_sequence_lengths <- getLength(myloasm_contig)
# Convert to DataFrame
myloasm_length_data <- data.frame(Length = myloasm_sequence_lengths) %>%
dplyr::mutate(tool = "myloasm")
# concat DataFrame
length_data <- dplyr::bind_rows(myloasm_length_data)
# Density plot
g <- ggplot(length_data, aes(x = Length, fill = tool, color = tool)) +
geom_density(alpha=0.5) +
ylim(c(0, 4.5e-05)) +
xlim(c(0, 2.0e+5)) +
labs(title = "Distribution of contig length", x = "Contig Length", y = "Freqency")
ggsave(file = "myloasm.png", plot = g)

fastaヘッダーの命名規則は下記のとおりです。
grep ">" myloasm_results/pac/assembly_primary.fa | head -n 1
>u550136ctg_len-223999_circular-no_depth-4-3-3_duplicated-no mult=1.00
u550136ctgは識別子で、len-は塩基配列長です。
circular-は環状化の判定結果(yes/no)で、コンティグがアセンブリグラフに自己ループを持ち、下記の場合にyesとなるそうです。
- 平均depthがx5以上
- 単一nodeと単一Edgeで構成されている
カウントしてみました。
# 環状化
apps=(nanopore pac)
for app in ${apps}; do
echo "${app}"
grep ">" myloasm_results/${app}/assembly_primary.fa | awk -F'_' '{if($3=="circular-yes"){print $3}}' | wc -l
done
nanopore 20 (/2048)
pac 139 (/14626)
seqkit/BBtoolsのcontig数の結果を考えると1%程度のcontigが環状化出来ている判定ですね。
また、自己ループしてるが、(1)または(2)を満たさない場合、circular-possiblyと表現されます。
grep "possibly" myloasm_results/pac/assembly_primary.fa | head -n 1
>u1108930ctg_len-3438462_circular-possibly_depth-9-7-6_duplicated-no mult=1.00
# 準環状化
apps=(nanopore pac)
for app in ${apps}; do
echo "${app}"
grep ">" myloasm_results/${app}/assembly_primary.fa | awk -F'_' '{if($3=="circular-possibly"){print $3}}' | wc -l
done
nanopore 48 (/2048)
pac 94 (/14626)
これはもちろんのこと。

>u106493ctg_len-2151012_circular-yes_depth-26-26-25_duplicated-no mult=1.00
これも環状判定らしい。

>u70445ctg_len-1308695_circular-yes_depth-16-16-16_duplicated-no mult=1.00
準環状判定のコンティグは以下のようなやつ。たしかになんかひっついてる。

>u178337ctg_len-1781649_circular-possibly_depth-7-7-6_duplicated-no mult=1.00
Depthはdepth-X1-X2-X3の形式で表され、3つ値が記載されますが、ヌクレオチド同一性の各閾値ごとの値を表しています。
- X1: 約99%を超える真のヌクレオチド類似性 (myloasmのSNP+k-mer定式化を使用して推定) の配置を可能にするDepthです
- X2: 99.75%を超える類似度のDepthです
- X3: 100%の類似性のDepthです
K-merの多重性と重複
mult=Xで表され、これは繰り返し性の推定割合であり、品質管理に役立ちます。
条件として、コンティグ全体で21-merをカウントし、最も頻度の高いk-merと最も頻度の低いk-merの10%を削除した後、それらが平均してどの程度の頻度で繰り返されるかを確認することによって推定されます。
- 最も完全な原核生物ゲノムには、 mult-1.00が含まれているはずです
- myloasmは、2つの類似株のゲノムを誤って結合することがあります。この場合、multは1.0を超えており、コンタミネーションを示しています
- ロングリードには重複したプラスミドが含まれることもよくあります。場合によっては、プラスミドが誤って重複したり、誤って環状化したりして、mult1を超える値になることがあります
multが>1.1の場合はduplicated(重複)がpossiblyと表され、>1.5の場合はyesと表現されます。それ以外のはnoです。
アセンブリグラフ: final_contig_graph.gfa
結果フォルダ内のfinal_contig_graph.gfaは、GFA形式のアセンブリグラフファイルです。Bandageで視覚化可能です。
Bandage load myloasm_results/pac/final_contig_graph.gfa
Bandage load myloasm_results/nanopore/final_contig_graph.gfa
USAGE Bandage
____ _
| _ \ | |
| |_) | __ _ _ __ __| | __ _ __ _ ___
| _ < / _` | '_ \ / _` |/ _` |/ _` |/ _ \
| |_) | (_| | | | | (_| | (_| | (_| | __/
|____/ \__,_|_| |_|\__,_|\__,_|\__, |\___|
__/ |
|___/
Version: 0.9.0
Usage: Bandage <command> [options]
Commands: <blank> Launch the Bandage GUI
load Launch the Bandage GUI and load a graph file
info Display information about a graph
image Generate an image file of a graph
querypaths Output graph paths for BLAST queries
reduce Save a subgraph of a larger graph
Options: --help View this help message
--helpall View all command line settings
--version View Bandage version number
Online Bandage help: https://github.com/rrwick/Bandage/wiki
- PacBio

- Nanopore

データボリュームによってだいぶ結果が違うようで、PacBioのデータはgut microbiomeですが、NanoporeのデータはOral microbiomeです。
データQC
アセンブリしたのでその評価を実施します。myloasmのQCスクリプトセットであるmylotoolsを使用します。
mylotoolsによるデータQC
以下のコマンドでインストール出来ます。
# 仮想環境にはActivateしておく
mamba activate myloasm_env
# Clone
git clone https://github.com/bluenote-1577/mylotools
cd mylotools
pip install .
cd ..
確認
mylotools --help
usage: mylotools [-h] [--version] {plot,strain-viz,extract-contigs} ...
mylotools - utility tools for myloasm
positional arguments:
{plot,strain-viz,extract-contigs}
plot Generate a plot of various statistics for one of myloasm's output contigs
strain-viz Visualize overlaps between and within two or more similar contigs
extract-contigs Extract contigs > X bp into a folder as its own fasta
options:
-h, --help show this help message and exit
--version show program's version number and exit
まず、長い(>300kb)誤った原核生物コンティグを除外する最も簡単な方法は、個々のコンティグに対してCheckM2を実行することです。キメラコンティグが十分に長い場合、複数の生物からの重複したマーカー遺伝子が存在するため、CheckMのcontamination値が高くなることが想定されます。
長さ>= Xbpのすべてのコンティグを抽出してCheckM2を実行するには、次のようにします。抽出するcontig長で>300kbを超えるものがなかったので>3kbとしました。
# PacBio
cd myloasm_results/pac/
mylotools extract-contigs --output-folder mylotools_results --min-contig-length 3000000
# Nanopore
cd myloasm_results/nanopore/
mylotools extract-contigs --output-folder mylotools_results --min-contig-length 3000000
CheckM2の実行
checkm2 predict --input mylotools_results -x fa -o checkm2_results --threads 32
環状化判定されているもの対象にして結果を見てみます。
# PacBioのデータ
grep -i "circular-yes" checkm2_results/quality_report.tsv
u1129721ctg_len-3075712_circular-yes_depth-35-27-25_duplicated-no 100.0 0.98 Neural Network (Specific Model) 11 0.837 3075712 314.6459400146306 3075712 0.55 27341 3075712 None
u1355330ctg_len-3446150_circular-yes_depth-8-6-5_duplicated-no 100.0 0.26 Neural Network (Specific Model) 11 0.869 3446150 326.98496732026143 3446150 0.42 3060 1 3446150 None
u1398258ctg_len-3484450_circular-yes_depth-18-15-15_duplicated-no 99.99 0.78 Neural Network (Specific Model) 11 0.859 3484450 306.69131635471 3484450 0.56 3259 1 3484450 None
u1426794ctg_len-4971160_circular-yes_depth-42-31-29_duplicated-no 100.0 0.49 Neural Network (Specific Model) 11 0.903 4971160 355.0154028436019 4971160 0.46 42201 4971160 None
u1510879ctg_len-3393118_circular-yes_depth-11-10-10_duplicated-no 96.83 1.57 Gradient Boost (General Model) 11 0.864 3393118 344.76259246213453 3393118 0.57 28391 3393118 None
u1530132ctg_len-3733716_circular-yes_depth-8-8-8_duplicated-no 99.98 0.31 Neural Network (Specific Model) 11 0.894 3733716 381.0875512995896 3733716 0.4 2924 1 3733716 None
u1565868ctg_len-3254961_circular-yes_depth-24-21-20_duplicated-no 99.85 2.54 Neural Network (Specific Model) 11 0.861 3254961 322.6208204067563 3254961 0.58 29011 3254961 None
u1641272ctg_len-3503503_circular-yes_depth-37-26-25_duplicated-no 100.0 1.15 Neural Network (Specific Model) 11 0.868 3503503 323.0722469764481 3503503 0.44 31421 3503503 None
u1704344ctg_len-3058751_circular-yes_depth-15-15-14_duplicated-no 99.96 0.49 Neural Network (Specific Model) 11 0.863 3058751 311.53960396039605 3058751 0.57 28281 3058751 None
u313000ctg_len-3222425_circular-yes_depth-31-29-28_duplicated-no 99.98 1.13 Neural Network (Specific Model) 11 0.87 3222425 308.92868933641466 3222425 0.56 30291 3222425 None
u37492ctg_len-3088247_circular-yes_depth-22-21-20_duplicated-no 99.98 3.86 Neural Network (Specific Model) 11 0.872 3088247 314.61756473058085 3088247 0.42 2858 1 3088247 None
u383992ctg_len-4945658_circular-yes_depth-37-30-28_duplicated-no 100.0 3.26 Neural Network (Specific Model) 11 0.863 4945658 313.8094190140845 4945658 0.43 45441 4945658 None
u386162ctg_len-3173188_circular-yes_depth-45-43-42_duplicated-no 98.87 1.32 Neural Network (Specific Model) 11 0.885 3173188 338.52926300578036 3173188 0.44 27681 3173188 None
u647030ctg_len-3342461_circular-yes_depth-76-63-60_duplicated-no 100.0 0.21 Neural Network (Specific Model) 11 0.875 3342461 311.6725183530163 3342461 0.41 31331 3342461 None
u71463ctg_len-4489393_circular-yes_depth-88-70-63_duplicated-no 99.98 0.51 Neural Network (Specific Model) 11 0.858 4489393 308.26653883029724 4489393 0.41 4172 1 4489393 None
u897218ctg_len-3685032_circular-yes_depth-35-35-35_duplicated-no 99.97 0.11 Neural Network (Specific Model) 11 0.836 3685032 355.2297857636489 3685032 0.45 28941 3685032 None
u934176ctg_len-4413985_circular-yes_depth-18-18-17_duplicated-no 99.89 0.49 Neural Network (Specific Model) 11 0.902 4413985 356.0474022495983 4413985 0.46 37341 4413985 None
2列目がCompleteness %で3列目がComtamination %です。5%以下のものばかりで着目するようなものがなかったですね。ただ、myloasmの環状化判定とCheckM2のCompleteness %が関連していることの確認が出来ました。
一応、原核生物のゲノムの場合、CheckM2のCompleteness %が低いと環状化が未完で有ることが示唆されます。
ただし、
- 完全な細胞小器官ゲノム(ミトコンドリア、微小真核生物のプラスチド)は、CheckM2の完全性(> 30%、<90%)が重要ではない
- 二次染色体や複数の染色体を持つゲノムは完全性が低い可能性がある
- 一部の微生物系統は、完全であってもCheckM2 Completeness %が低い
myloasmのcircular-possiblyタグは、信頼性の低い環状ゲノム(カバレッジが低いかアセンブリグラフがあいまい)であることを示しますが、CheckM2スコアが良好な場合は、完全なゲノムを復元できている可能性が高いです。
重複/系統キメラに対するk-mer多重度統計を使用したQC
Myloasmのfasta出力には、コンティグ内で21merが繰り返される頻度(多重度)に関する情報が含まれています。作者の経験では、原核生物のcontigの平均k-mer多重度はほぼ常に1.00に近くなるとのことです。
非常に長いコンティグ(1M bp以上)で多重度が1.05を超える場合は、同一種の複数の系統から生じたキメラである可能性があります。またはdeplicated-にyes/possiblyが設定された場合には多重度が高くなります。
また、小さなゲノム(例:ウイルス)の場合、期待されるk-mer多重度は1.00から逸脱する可能性があります。
しかし、小さなコンティグでk-mer多重度が1より大きい場合には結果を疑う必要がある。また、k-mer多重度が 2, 3か整数倍のコンティグは、重複したコンティグを示している可能性があります。
mylotoolsによる手動調査
mylotools plotでロングリードオーバーラップを手動で検査しキメラ配列を調査します。
GC含有量やオーバーラップ read、コンティグ全体のカバレッジ情報を取得するために、mylotools plotと呼ばれるプロットユーティリティコマンドが提供されています。
例えば、関心のあるコンティグ(u1129721ctg)に対して次のように実行します。
# you should be in the results directory
cd myloasm_results
mylotools plot u1129721ctg
出力されたu1129721ctg_analysis.htmlは下記のようなグラフが表示されているはずです。

上記は説明に適したデータではないので、開発者のチュートリアルの図を例に結果をの確認を進めます。
嫌気性消化槽のメタゲノムから取得されたCheckM2のCompleteness 99%である一方、Contamination %が18%のコンティグです。2.6Mbp付近に不規則性が確認できます。

https://myloasm-docs.github.io/assets/myloplot-bp.png
- パネル1: GC含有量
window全体で平均されたGC %グラフです。原核生物ゲノムは、ゲノム全体にわたってGC含量が比較的一定であるはずです。GC含量に急激な変化が見られる場合は、キメラ切断点を示唆している可能性があります。 - パネル2: 累積のGC Skew
GC Skewの詳細については、こちらの論文かこちらの論文を参照してください。多くの原核生物は、複製開始点と終了点付近にピークと谷があります。 - パネル3: ゲノム全体のカバレッジ
各ドットは、コンティグを構築するリードを表しています。リードカバレッジには3つの異なる値があり、DP1は最も許容度の高いカバレッジ値で、DP3は正確なカバレッジ(完全なアライメント)を示します。
詳細はこちらを参照下さい。 - パネル4: アセンブリグラフ内の重複情報
各ドットはリードオーバーラップを表しています。配列長とリードIDに関する統計情報が表示されます。色はリード間で共有されている/異なるSNPの数を示します。理想的なコンティグではリード間で異なるSNPは存在しませんが、場合によってはそれに準拠しない場合もあります(例えば、組換えレベルが高いなど)。
作者の図では以下の点に着目しています。
- GC含有量は顕著に減少している
- 累積GCスキューは再び上昇し、下降する
- DP1/DP2のカバレッジは変動性が高く、ブレークポイント前のDP1/DP2は一貫して低い。
これらはキメラコンティグである強い証拠です。
2.6Mbp以降の塩基を除去してCheckM2を再実行したところ、Completeness 97%で、コンタミネーションが0%のコンティグが得られました。
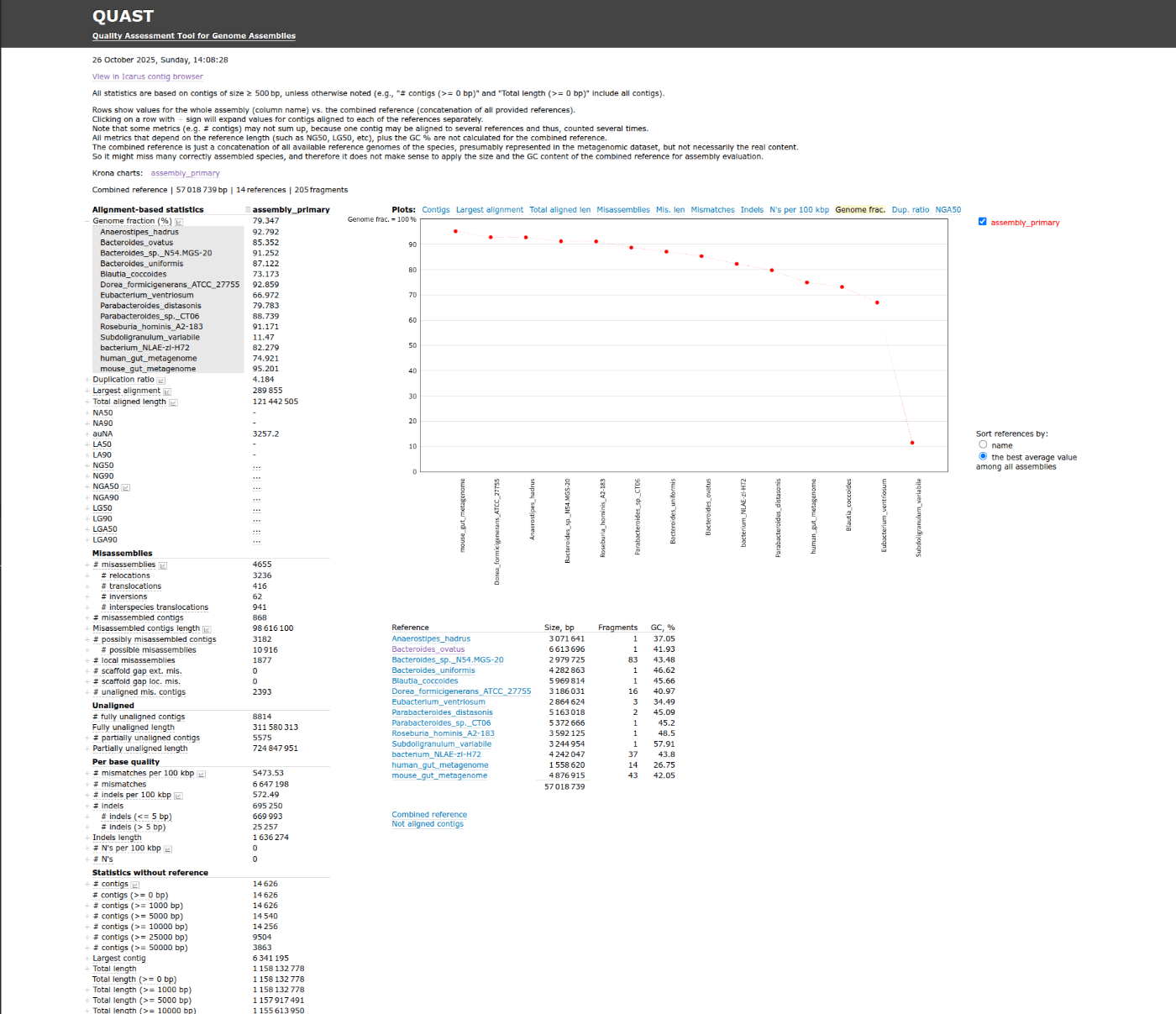
metaQUAST
PacBioのデータで実行してみます。リファレンスはないので16sのBlastnの結果より、リファレンスゲノムがNCBIからダウンロードされて比較に使用されます。
metaquast.py myloasm_results/pac/assembly_primary.fa --threads 32
1時間くらいで終了しました。
ls quast_results/latest/ -1
combined_reference
icarus.html
icarus_viewers
krona_charts
metaquast.log
not_aligned
quast_downloaded_references
report.html
runs_per_reference
summary
report.htmlから各ページに飛ぶことが出来ます。また、フォルダ内にあるファイルからも同様の結果を確認可能です。
- QUAST report html
- Krona plot

- Icarus contig browser

metaQUASTを実行したことがなかったので解析かけましたが、あくまでメタゲノムデータから取得したcontigを使ったゲノム比較なので、この段階でのmetaQUASTの結果の有用性は低いかもしれません。
関連記事
- Autocyclerの著者のブログ: https://rrwick.github.io/2025/09/23/autocycler-benchmark-update.html
- myloasmのv0.2.0の結果が優秀だったのでAutocyclerに追加する予定の模様
Discussion