LRBinner: ロングリードメタゲノミクスのデータ組成とカバレッジ情報を同時に利用してビニングする
LRBinner
メタゲノミクスにおけるビニングステップは微生物群集の特徴付けにおける重要なステップです。
Illuminaシーケンサーなどの第2世代のショートリードシーケンサーは、生成されるリード単体の情報が限られているため、一般的にはメタゲノムアセンブラーによってアセンブルを実行します。
一方、第2世代シーケンサーより長鎖の配列を生成する第3世代のシーケンサーは、ショートリードのアセンブル後のcontig配列と同等以上の長さを持つRaw readを生成します。
LRBinnerの発表時の基本的なContigのビニングツールは、カバレッジ情報がなくエラー率の高いロングリードシーケンスには直接適用できないか、ロングリード用のビニングツールを利用したとしても、データの構成とカバレッジの情報を単独使用もしくは独立して両者を使用するに留まっていました。
これは、存在量の少ない種に対応するBinが無視されたり、不均一なカバレッジを持つ種に対応するBinが誤って分割されたりする可能性があります。
これらの課題に対応するため、LRBinnerではデータの構成とカバレッジ情報を組み合わせつつ、距離ヒストグラムベースのクラスタリングアルゴリズムを使用することで任意のクラスター (Bin) を生成します。

LRBinner Intro参照
Insatall LRBinner
依存関係のあるツールのインストール
mamba create -n lrbinner -y python=3.10 numpy scipy seaborn h5py hdbscan gcc openmp tqdm biopython fraggenescan hmmer tabulate pytorch pytorch-cuda=11.8 -c pytorch -c nvidia -c bioconda
LRBinnerをダウンロード
git clone https://github.com/anuradhawick/LRBinner.git
ビルドする。
cd LRBinner
python setup.py build
BUILD FINISHEDと出れば終了
USAGE LRBinner
help
python3 LRBinner/lrbinner.py -h
usage: lrbinner.py [-h] [--version] {reads,contigs} ...
LRBinner Help. A tool developed for binning of metagenomics long reads (PacBio/ONT) and long read assemblies. Tool utilizes
composition and coverage profiles of reads based on k-mer frequencies to perform dimension reduction via a deep variational
auto-encoder. Dimension reduced reads are then clustered. Minimum RAM requirement is 9GB (4GB GPU if cuda used).
options:
-h, --help show this help message and exit
--version, -v Show version.
LRBinner running Mode:
{reads,contigs}
reads for binning reads
contigs for binning contigs
readsモードのhelp
usage: lrbinner.py reads [-h] --reads-path READS_PATH [--k-size {3,4,5}] [--bin-size BIN_SIZE] [--bin-count BIN_COUNT]
[--ae-epochs AE_EPOCHS] [--ae-dims AE_DIMS] [--ae-hidden AE_HIDDEN] [--threads THREADS]
[--separate] [--cuda] [--resume] --output <DEST> [--min-bin-size MIN_BIN_SIZE]
[--bin-iterations BIN_ITERATIONS]
options:
-h, --help show this help message and exit
--reads-path READS_PATH, -r READS_PATH
Reads path for binning
--k-size {3,4,5}, -k {3,4,5}
k value for k-mer frequency vector. Choose between 3 and 5.
--bin-size BIN_SIZE, -bs BIN_SIZE
Bin size for the coverage histogram.
--bin-count BIN_COUNT, -bc BIN_COUNT
Number of bins for the coverage histogram.
--ae-epochs AE_EPOCHS
Epochs for the auto_encoder.
--ae-dims AE_DIMS Size of the latent dimension.
--ae-hidden AE_HIDDEN
Hidden layer sizes eg: 128,128
--threads THREADS, -t THREADS
Thread count for computations
--separate, -sep Flag to separate reads/contigs into bins detected. Avaialbe in folder named 'binned'.
--cuda Whether to use CUDA if available.
--resume Continue from the last step or the binning step (which ever comes first). Can save time needed count
k-mers.
--output <DEST>, -o <DEST>
Output directory
--min-bin-size MIN_BIN_SIZE, -mbs MIN_BIN_SIZE
The minimum number of reads a bin should have.
--bin-iterations BIN_ITERATIONS, -bit BIN_ITERATIONS
Number of iterations for cluster search. Use 0 for exhaustive search.
contigモードのhelp
usage: lrbinner.py contigs [-h] --reads-path READS_PATH [--k-size {3,4,5}] [--bin-size BIN_SIZE] [--bin-count BIN_COUNT]
[--ae-epochs AE_EPOCHS] [--ae-dims AE_DIMS] [--ae-hidden AE_HIDDEN] [--threads THREADS]
[--separate] [--cuda] [--resume] --output <DEST> --contigs CONTIGS
options:
-h, --help show this help message and exit
--reads-path READS_PATH, -r READS_PATH
Reads path for binning
--k-size {3,4,5}, -k {3,4,5}
k value for k-mer frequency vector. Choose between 3 and 5.
--bin-size BIN_SIZE, -bs BIN_SIZE
Bin size for the coverage histogram.
--bin-count BIN_COUNT, -bc BIN_COUNT
Number of bins for the coverage histogram.
--ae-epochs AE_EPOCHS
Epochs for the auto_encoder.
--ae-dims AE_DIMS Size of the latent dimension.
--ae-hidden AE_HIDDEN
Hidden layer sizes eg: 128,128
--threads THREADS, -t THREADS
Thread count for computations
--separate, -sep Flag to separate reads/contigs into bins detected. Avaialbe in folder named 'binned'.
--cuda Whether to use CUDA if available.
--resume Continue from the last step or the binning step (which ever comes first). Can save time needed count
k-mers.
--output <DEST>, -o <DEST>
Output directory
--contigs CONTIGS, -c CONTIGS
Contigs path
LRBinnerの実行
githubリポジトリにあるテストデータのlink先からfastaファイルをダウンロードできなかったのでPacBioが公開しているヒトの腸内細菌データm64011_210224_000525を使用する。
PacBioのクラウドデータベースからデータを取得。
aria2c -x 16 https://downloads.pacbcloud.com/public/dataset/Sequel-IIe-202104/metagenomics/m64011_210224_000525.hifi_reads.fastq.tar.gz
tar -xvf metagenomics/m64011_210224_000525.hifi_reads.fastq.tar.gz
LRBinnerの実行
圧縮されたファイルは非対応なので.fastq, .fastaの形式で与える。kmer sizeで結果が変わるようなので-kを変えて3,4,5の3パターンでbinを生成します。以下は-k = 3のパターン。
python3 LRBinner/lrbinner.py reads -r m64011_210224_000525.hifi_reads.fastq -bc 10 -bs 32 -o lrb-k3 --resume --cuda -mbs 5000 --ae-dims 4 --ae-epochs 200 -bit 0 -k 3 -t 32
上記スクリプトを実行した場合、配列情報などは出力されません。-sepオプションをつけて実行するとbinned_readsというフォルダにBin-X.fastaが出力されます。
python3 LRBinner/separate_reads.py --reads m64011_210224_000525.hifi_reads.fastq -b lrb-k3/binning_result.pkl -o lrb-k3/bin/
生成されたbinのstatsを確認。
for i in {3..5}; do seqkit stats -a lrb-k${i}/bin/*/* ; done
- -k3 合計 8bin
| file | num_seqs | sum_len | min_len | avg_len | max_len | N50 |
|---|---|---|---|---|---|---|
| lrb-k3/bin/bin-0/reads.fasta | 377887 | 3927212660 | 450 | 10392.6 | 45570 | 11563 |
| lrb-k3/bin/bin-1/reads.fasta | 111607 | 1180797513 | 61 | 10580 | 42867 | 11823 |
| lrb-k3/bin/bin-2/reads.fasta | 427855 | 4461579941 | 89 | 10427.8 | 49376 | 11688 |
| lrb-k3/bin/bin-3/reads.fasta | 254828 | 2595221547 | 178 | 10184.2 | 39900 | 11126 |
| lrb-k3/bin/bin-4/reads.fasta | 239667 | 2425393628 | 238 | 10119.8 | 39607 | 11125 |
| lrb-k3/bin/bin-5/reads.fasta | 222318 | 2285313274 | 85 | 10279.5 | 38290 | 11391 |
| lrb-k3/bin/bin-6/reads.fasta | 130842 | 1323968759 | 267 | 10118.8 | 40865 | 11200 |
| lrb-k3/bin/bin-7/reads.fasta | 27142 | 290933733 | 1290 | 10718.9 | 36108 | 11978 |
- -k4 合計 19bin
| file | num_seqs | sum_len | min_len | avg_len | max_len | N50 |
|---|---|---|---|---|---|---|
| lrb-k4/bin/bin-0/reads.fasta | 106584 | 1127643708 | 61 | 10579.9 | 42867 | 11809 |
| lrb-k4/bin/bin-10/reads.fasta | 37527 | 399109988 | 1290 | 10635.3 | 36108 | 11912 |
| lrb-k4/bin/bin-11/reads.fasta | 185438 | 1869499602 | 238 | 10081.5 | 37223 | 11062 |
| lrb-k4/bin/bin-12/reads.fasta | 57796 | 586592891 | 300 | 10149.4 | 33091 | 11115 |
| lrb-k4/bin/bin-13/reads.fasta | 39214 | 383501750 | 572 | 9779.7 | 39132 | 10675 |
| lrb-k4/bin/bin-14/reads.fasta | 205934 | 2089615717 | 255 | 10147 | 39607 | 11211 |
| lrb-k4/bin/bin-15/reads.fasta | 18314 | 186640991 | 1413 | 10191.2 | 32321 | 11165 |
| lrb-k4/bin/bin-16/reads.fasta | 10966 | 108191051 | 617 | 9866 | 31875 | 11095 |
| lrb-k4/bin/bin-17/reads.fasta | 33837 | 349176539 | 669 | 10319.4 | 32985 | 11528 |
| lrb-k4/bin/bin-18/reads.fasta | 5367 | 56025029 | 897 | 10438.8 | 33879 | 11703 |
| lrb-k4/bin/bin-1/reads.fasta | 15545 | 161304820 | 1592 | 10376.6 | 30893 | 11486 |
| lrb-k4/bin/bin-2/reads.fasta | 15702 | 172760739 | 1239 | 11002.5 | 34820 | 12325 |
| lrb-k4/bin/bin-3/reads.fasta | 48534 | 492468119 | 310 | 10146.9 | 45570 | 11243 |
| lrb-k4/bin/bin-4/reads.fasta | 16272 | 168493091 | 652 | 10354.8 | 33792 | 11468 |
| lrb-k4/bin/bin-5/reads.fasta | 456610 | 4735739815 | 368 | 10371.5 | 41239 | 11549 |
| lrb-k4/bin/bin-6/reads.fasta | 57318 | 599039597 | 549 | 10451.2 | 39900 | 11443 |
| lrb-k4/bin/bin-7/reads.fasta | 220136 | 2273328260 | 85 | 10326.9 | 49376 | 11612 |
| lrb-k4/bin/bin-8/reads.fasta | 74324 | 768259215 | 178 | 10336.6 | 37105 | 11272 |
| lrb-k4/bin/bin-9/reads.fasta | 186728 | 1963030133 | 267 | 10512.8 | 35920 | 11684 |
- -k5 合計 17bin
| file | num_seqs | sum_len | min_len | avg_len | max_len | N50 |
|---|---|---|---|---|---|---|
| lrb-k5/bin/bin-0/reads.fasta | 31120 | 332936497 | 1290 | 10698.5 | 36108 | 11965 |
| lrb-k5/bin/bin-10/reads.fasta | 462881 | 4799795226 | 85 | 10369.4 | 45570 | 11545 |
| lrb-k5/bin/bin-11/reads.fasta | 301265 | 3128461141 | 61 | 10384.4 | 49376 | 11664 |
| lrb-k5/bin/bin-12/reads.fasta | 16021 | 161640844 | 358 | 10089.3 | 33562 | 10975 |
| lrb-k5/bin/bin-13/reads.fasta | 44302 | 446935124 | 238 | 10088.4 | 35294 | 11083 |
| lrb-k5/bin/bin-14/reads.fasta | 25897 | 260723135 | 846 | 10067.7 | 38436 | 11172 |
| lrb-k5/bin/bin-15/reads.fasta | 13238 | 132234765 | 288 | 9989 | 32180 | 11239 |
| lrb-k5/bin/bin-16/reads.fasta | 110103 | 1137595052 | 414 | 10332.1 | 39607 | 11426 |
| lrb-k5/bin/bin-1/reads.fasta | 15744 | 163167364 | 572 | 10363.8 | 30893 | 11475 |
| lrb-k5/bin/bin-2/reads.fasta | 104463 | 1108553075 | 255 | 10611.9 | 42867 | 11860 |
| lrb-k5/bin/bin-3/reads.fasta | 58656 | 612653206 | 549 | 10444.9 | 39900 | 11433 |
| lrb-k5/bin/bin-4/reads.fasta | 48414 | 493758499 | 310 | 10198.7 | 40865 | 11303 |
| lrb-k5/bin/bin-5/reads.fasta | 26274 | 271888628 | 274 | 10348.2 | 34997 | 11496 |
| lrb-k5/bin/bin-6/reads.fasta | 149656 | 1532767339 | 267 | 10241.9 | 37777 | 11308 |
| lrb-k5/bin/bin-7/reads.fasta | 182000 | 1836579609 | 178 | 10091.1 | 39132 | 11031 |
| lrb-k5/bin/bin-8/reads.fasta | 104797 | 1044214495 | 243 | 9964.2 | 37223 | 10892 |
| lrb-k5/bin/bin-9/reads.fasta | 97315 | 1026517056 | 902 | 10548.4 | 32589 | 11747 |
それぞれの要素をplot
python script for violin plot
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
# set using header info
header = ['file', 'num_seqs', 'min_len', 'avg_len', 'max_len']
# read stats
dat_k3 = pd.read_table("3.stats", usecols= header, sep = '\t', index_col = None)
dat_k3['cate'] = 'k3'
dat_k4 = pd.read_table("4.stats", usecols= header, sep = '\t', index_col = None)
dat_k4['cate'] = 'k4'
dat_k5 = pd.read_table("5.stats", usecols= header, sep = '\t', index_col = None)
dat_k5['cate'] = 'k5'
# concat dat
merged_dat = pd.concat([dat_k3,dat_k4,dat_k5], ignore_index= True)
merged_dat.head(10)
# sequence num violin plot
sns.violinplot(data = merged_dat, x = 'cate', y = 'num_seqs',color="0.8")
sns.stripplot(data = merged_dat, x = 'cate', y = 'num_seqs',jitter=True, zorder=1)
plt.show()
# total sequence length plot
sns.violinplot(data = merged_dat, x = 'cate', y = 'sum_len',color="0.8")
sns.stripplot(data = merged_dat, x = 'cate', y = 'sum_len',jitter=True, zorder=1)
plt.show()
# max len violin plot
sns.violinplot(data = merged_dat, x = 'cate', y = 'max_len',color="0.8")
sns.stripplot(data = merged_dat, x = 'cate', y = 'max_len',jitter=True, zorder=1)
plt.show()
# min len violin plot
sns.violinplot(data = merged_dat, x = 'cate', y = 'min_len',color="0.8")
sns.stripplot(data = merged_dat, x = 'cate', y = 'min_len',jitter=True, zorder=1)
plt.show()
- num sequence
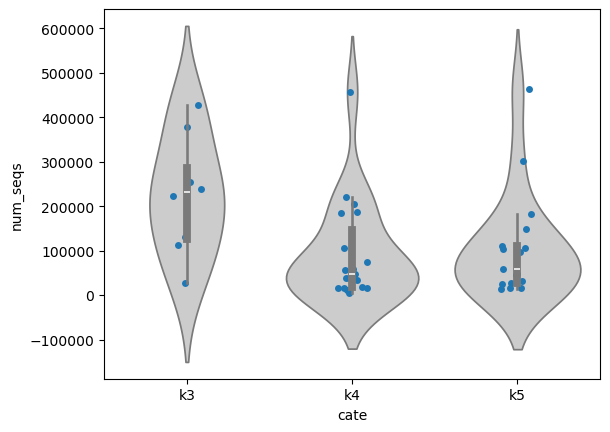
- total sequence length

- max length
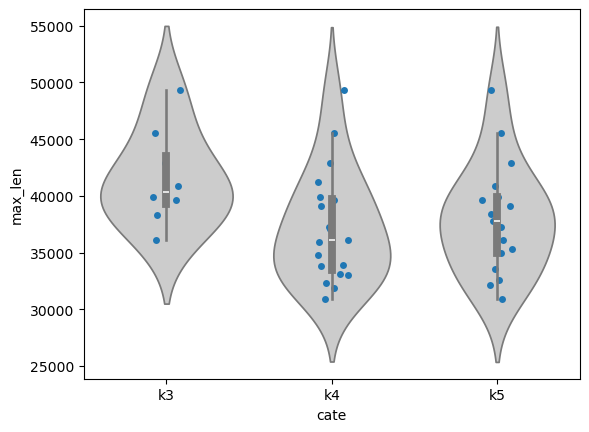
- min length
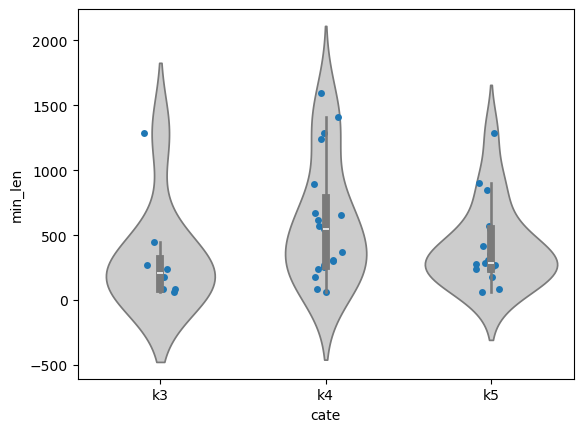
他のツールに比べてBinの数が少ないような。inputにPacBioのRawfastqを与えましたが、hifiasm-metaでアセンブルした後のcontig.fastaで実行しても大差ありませんでした。
SemiBin2での論文でも正しい結果を生成出来ていなみたいなので今後の改良に期待。
Information
- 自作PC1
- libraries
Name Version Build Channel numpy 1.26.3 py310hb13e2d6_0 conda-forge scipy 1.12.0 py310hb13e2d6_2 conda-forge seaborn 0.13.2 hd8ed1ab_0 conda-forge h5py 3.10.0 nompi_py310h65828d5_101 conda-forge pytorch 2.2.0 py3.10_cuda11.8_cudnn8.7.0_0 pytorch pytorch-cuda 11.8 h7e8668a_5 pytorch pytorch-mutex 1.0 cuda pytorch torchtriton 2.2.0 py310 pytorch fraggenescan 1.31 h031d066_6 bioconda hmmer 3.4 hdbdd923_0 bioconda hdbscan 0.8.33 py310h1f7b6fc_4 conda-forge
Discussion