4. クオリティチェックとフィルタリング
前回はこちら。
バイオリンプロットを見ながら、シングルセルデータのクオリティを確認するとともに、状態の良くない細胞のデータを除外(フィルタリング)する方法を紹介します。
4.1 クオリティチェック
検出されている遺伝子の数が極端に少ない細胞もあります。バイオリンの下のほうに位置しているドットが該当します。数十個の遺伝子しか検出できていない細胞は、データとしてはクオリティが良くないと考えられ、後述するフィルタリングのステップで除外することになります。
また、クオリティをチェックする指標の1つとして、「ミトコンドリア由来の遺伝子の検出された遺伝子に占める割合(percent.mt)」があります。
チュートリアルにおいて、PercentageFeaturesSet()関数を用いて算出されている部分が該当します。この処理により、Seurat オブジェクト内の percent.mt という変数に、各細胞におけるミトコンドリア遺伝子の割合が格納されます。
pbmc_raw[["percent.mt"]] <- PercentageFeatureSet(pbmc_raw, pattern = "^MT-")
この percent.mt と共に、nFeature_RNA, nCount_RNA をバイオリンプロットで表示したものが下記のプロットです。引数の features にまとめて指定することができます。
VlnPlot(pbmc_raw, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
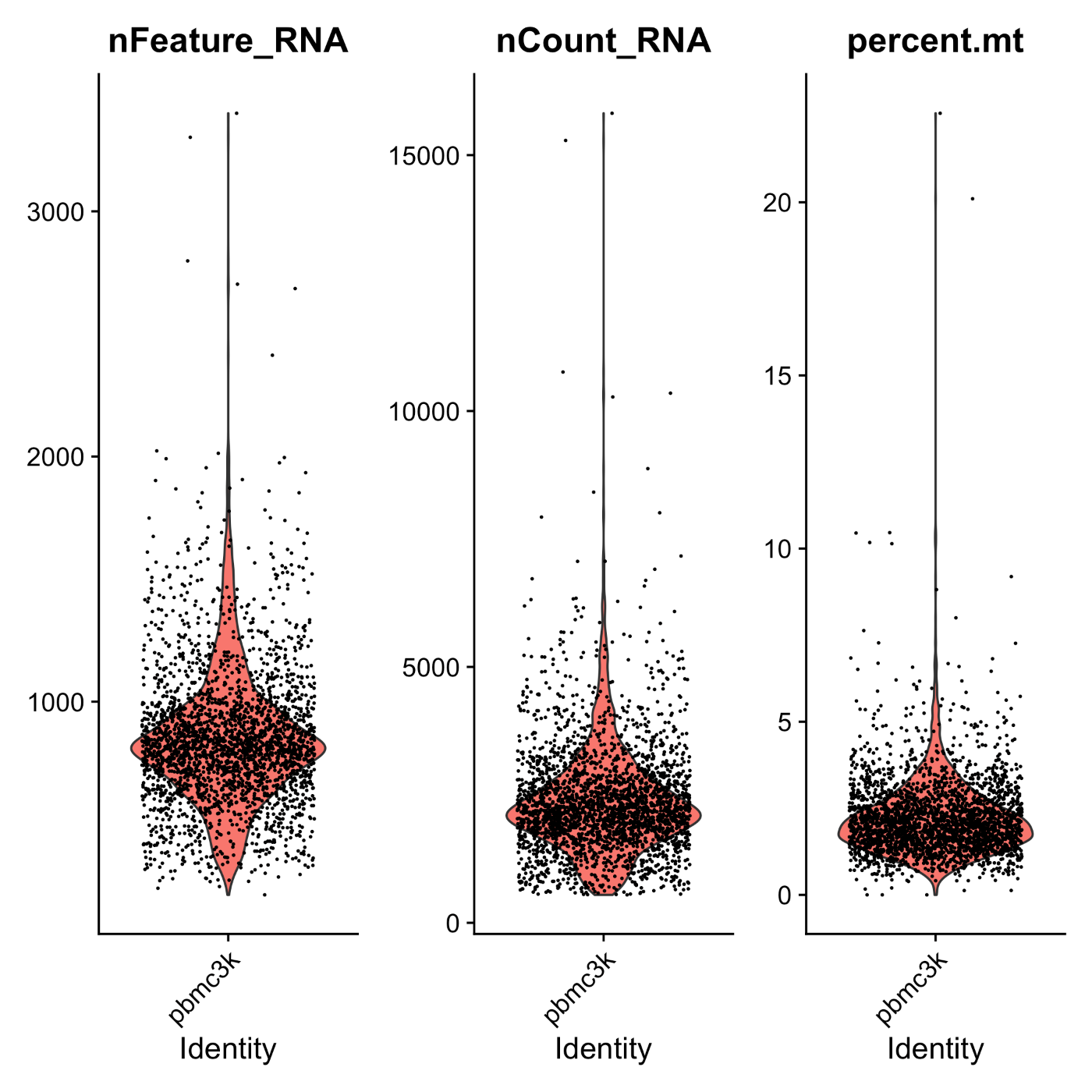
左の nFeature_RNA は、前回 紹介しました。真ん中の nCount_RNA は、各細胞におけるカウントされたリード数の合計を示しています。1細胞あたり2000リードほどが検出されていることが分かります。nFeature_RNA に示されるように検出された遺伝子は、たかだか数千個ですから、ほとんどの遺伝子は、リードが数本しか検出されていないと考えられます。このようにシングルセルのデータは、1細胞で考えると薄いデータです。
一番、右の percent.mt を確認すると、ほとんどの細胞では、ミトコンドリア由来の遺伝子は、5%未満となっています。これは比較的良好なデータと考えられます。一般的に、死細胞ではミトコンドリア由来の遺伝子の割合が高くなるとされており、数十パーセントを示すような細胞は、次のフィルタリングのステップで、除外します。
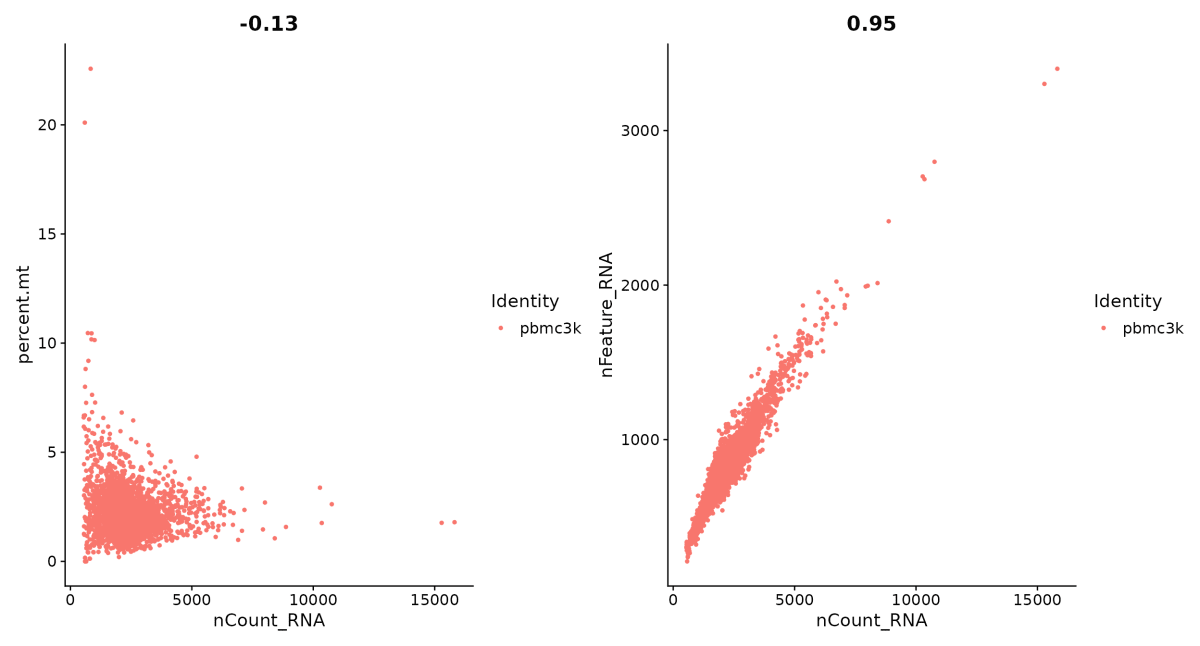
*特定の遺伝子だけ数万リードも検出されているような細胞は、イレギュラーと考えられます。チュートリアルでは、FeatureScatter() 関数を用いて散布図を作成することで、それらをチェックしています。1個の細胞に含まれるリード(nCount_RNA)が多いほど、検出される遺伝子数(nFeature_RNA)も多くなるのが予想されます。つまり、 ほとんどの細胞が、y = x 上に並び、これから外れているのは例外と考えられます(散布図右)。
4.2 フィルタリング
上記のステップでクオリティの良くない細胞が確認できた場合、それらを除外します(フィルタリング)。Seurat では、 subset()関数を使って、条件を満たす細胞だけを選択して、新しいデータセットを定義できます。
pbmc <- subset(pbmc_raw, subset = nFeature_RNA > 200 & percent.mt < 5)
関数名と引数が同じで分かりづらいですが、subset()関数の引数 subset に、フィルタリングの条件式を指定します。上記の nFeature_RNA > 200 & percent.mt < 5の部分が、その条件式です。つまり、「遺伝子数が 200以上」かつ「ミトコンドリア由来の遺伝子の割合が 5 %以下」を選択したことになります。
死細胞を取り除く条件は、5% 以下とする決まりはありません。状態の良くないデータセットによっては、5%では厳しすぎて、ほとんどの細胞が残らないこともあると思います。その場合は、10% から 20 % などに変更することになります。
* ここでは、 pbmc_raw に subset()関数を適用して、選択された結果を新しい pbmc というオブジェクトに格納しています。本家のチュートリアル では、subset()関数を適用するのが、同じ pbmc になっていますが、これはプログラムでよく行われる処理です。関数によって変更された内容を同じオブジェクトに格納することで、元のオブジェクトが上書きされることになります。
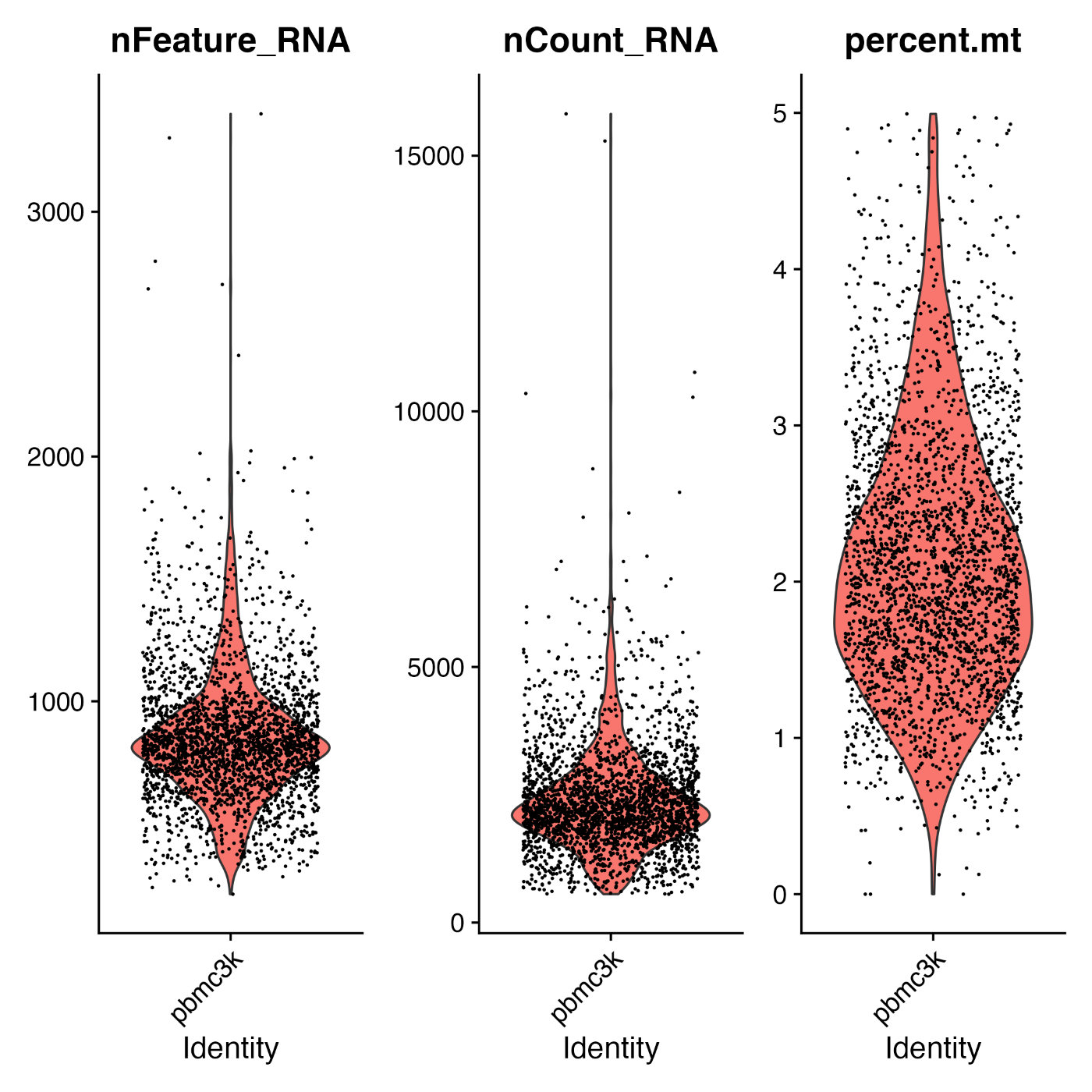
フィルタリングした結果を再度、バイオリンプロットで確認してみましょう。
下図のように、 percent.mt の上限が 5% となり、除外されていることが分かります。
フィルタリングしただけでは、前処理として、まだ不十分です。カウント値は細胞ごと、遺伝子ごとのスケールが違いすぎて、そのまま比較するには都合が悪いためです。次のステップとして、データの正規化の処理へ進みます。
Discussion