3. シングルセルデータの概略
前回はこちら。
例として、Seurat で読み込んだPBMC のデータを見ていきながら、シングルセルのデータはどのような形状のデータで、どんな特徴があるかを解説します。
3.1 シングルセルデータは、どんなデータ?
まず、チュートリアルでも紹介されている PBMC のデータを読み込んで Seurat オブジェクトを作成します。解説は、 前回 を参照してください。データはこちらからダウンロードできます。
library(tidyverse)
library(Seurat)
pbmc_data <- Read10X(data.dir = "./filtered_gene_bc_matrices")
pbmc_raw <- CreateSeuratObject(counts = pbmc_data, project = "pbmc3k",
min.cells = 3, min.features = 200)
pbmc_raw が読み込まれたカウント結果を Seurat オブジェクトに変換したものです。R のプロンプトにオブジェクト名の pbmc_raw を入力すると、簡単な説明が表示されます。
> pbmc_raw
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
この表示から、13,714 個の遺伝子 (=features) のデータが、2700細胞 (=samples) 含まれたオブジェクトであることが確認できます。
データの形状
シングルセルのデータは、1行につき1個の遺伝子、1列につき1つのバーコード(=細胞)から構成される行列と捉えることができます。この pbmc_raw のデータについては、13,724個の遺伝子が検出されていて、2700細胞ぶんのデータであるので、13,724行 x 2,700列の行列になるということです。実際に、Seurat オブジェクトの中には、この行列の形式で格納されています。
イメージしやすいように、例として、CD3D と CD3E の2つの遺伝子について、3細胞だけ表示させてみます。
> pbmc_raw$RNA@data[c("CD3D", "CD3E"), 1:3]
2 x 3 sparse Matrix of class "dgCMatrix"
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1
CD3D 4 . 10
CD3E 2 . 2
行の名前に遺伝子、列の名前にバーコードが表示されています。つまり、1列目の細胞については、CD3Dのリードが4本、CD3E のリードが2本、カウントされていたというこです。2列目の細胞については、2遺伝子ともドット(.)で表示されているので、カウントはゼロ(=未検出)ということです。
実際には、2700列のデータが横に並ぶことになります。このようにシングルセルのデータは、大きな行列の形状をしているため、たとえ、エクセルで開けたとしても、全体像を眺めることが難しいです。
そのため、シングルセルデータの全体像を確認するには、ボックスプロットや、バイオリンプロットを用いて視覚化することが通常です。
3.2 バイオリンプロットによる視覚化
Seurat には、読み込んだデータをバイオリンプロットで表示するための関数が備わっています。例のチュートリアルも、データのクオリティーチェック (QC: quality check) の方法として、紹介されています。バイオリンプロットを作成するには、VlnPlot() 関数を使用します。
バイオリンプロットで遺伝子数を確認
読み込んだデータをバイオリンプロットで表示する場合、どのパラメーターを視覚化したいのか引数で指定します。引数には、nFeatuer_RNA (=遺伝子数) や、 nCount_RNA (=リード数の合計) などが使用できます。
各細胞における遺伝子数を表示するには、下記のように指定します。
VlnPlot(pbmc_raw, features = c("nFeature_RNA"))
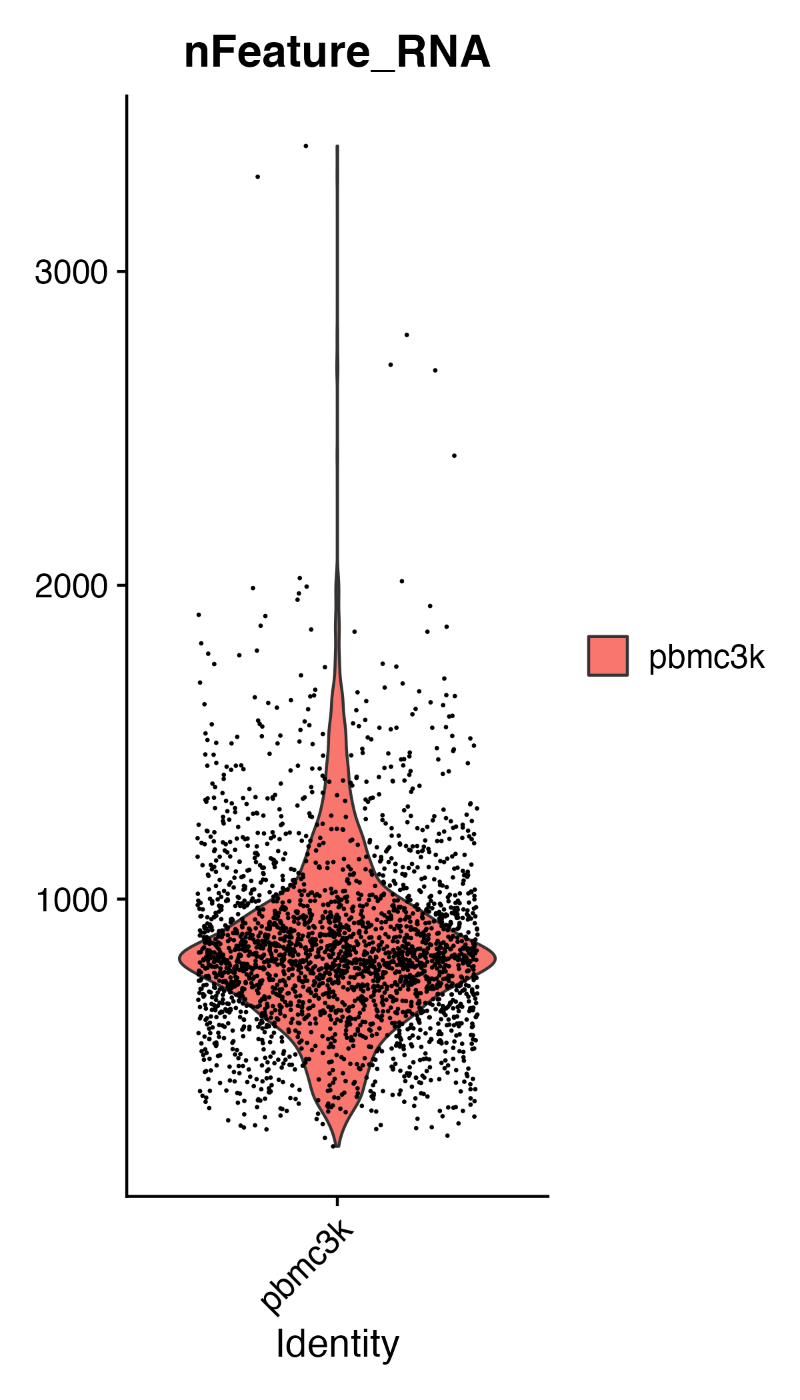
表示されるバイオリンプロットは、下図のようになります。
画像を保存するには、ggsave() が使えます。
ggsave("violin_plot.png", width = 4, height = 7, unit = "in")
バイオリンプロットに表示されている1個のドットが、1個の細胞を表しています。つまり、2700個のドットで、その細胞で検出された遺伝子の数が示されています。また、ドットの密度が背景の曲線の膨らみで示されています。(その形がバイオリンに見えるので、バイオリンプロット)
*なお、ドットの横の並びに意味はありません。ランダムで配置されているだけです。VlnPlot()を実行するたびに、ランダムな配置になります。
バイオリンプロットから確認できること
このプロットから、ほとんどの細胞では、検出されている遺伝子が 1,000 遺伝子前後であることが分かります。多いものでも、3,000 遺伝子を超える程度です。
つまり、シングルセルのデータでは、1細胞中に発現している遺伝子は、高々、数千遺伝子であり、ゲノム中の全ての遺伝子が検出されるわけではありません。
*これは「細胞1個あたりでは」の話です。サンプル全体では、前述のように、13,724個の遺伝子が検出されています。
このあたり、意外と誤解されていることも多いように思います。「RNA-seq だから精度が高く、1細胞のレベルで20,000遺伝子の全てが検出される」ようなイメージなのかもしれませんが、それは間違いです。精度が悪いと言っているのではありません。シーケンス量には限りがあるため、どうしても1細胞あたりのシーケンス量は薄くなり、検出される遺伝子数に限界があるという話です。
また、「発現している (express) 」ではなく、あえて、「検出された (detected)」と表現しているのも理由があります。シーケンサーはプロトコル上、「発現している遺伝子」を全て検出できるとは限りません。検出された遺伝子というのは、(いずれかの細胞で)少なくとも1本以上のリードがカウントされた遺伝子です。
逆に、検出されていない遺伝子は、リードがゼロということです。それも、今回は(その細胞では)ゼロだっただけです。「発現していない」ことの証明にはなりませんので注意してください。
Discussion