【化学でPython】ASE:分子シミュレーションをPythonでまるっと操作する
はじめに
この「化学でPython」シリーズでは、化学の分野で有用な Python ライブラリを紹介しています。
今回紹介するのは、ASE (Atomic Simulation Environment) です。
ASE とは?
原子・分子レベルのシミュレーションを行うための「司令塔」となるライブラリです。
分子や結晶の構築から、計算の実行、結果の解析までを統一的なインターフェースでまるっと操作できます。
インストール
pip で簡単にインストールできます。
pip install ase
基本的な使い方
まずは水分子を作成して、その原子の情報を表示する基本的な使い方を見てみましょう。
from ase import Atoms
from ase.build import molecule
# 水分子を作成
water = molecule('H2O')
# 原子の情報を表示
print(f"化学式: {water.get_chemical_formula()}")
print(f"原子数: {len(water)}")
print("各原子の位置 (Å):")
print(water.get_positions())
# 0番目の原子(酸素)の情報を取得
atom0 = water[0]
print(f"0番目の原子: {atom0.symbol}, 座標: {atom0.position}")
化学式: H2O
原子数: 3
各原子の位置 (Å):
[[ 0. 0. 0.119262]
[ 0. 0.763239 -0.477047]
[ 0. -0.763239 -0.477047]]
0番目の原子: O, 座標: [0. 0. 0.119262]
実践例: 金属表面への吸着エネルギー計算
実践的な例として、「銅 (Cu) の表面に金 (Au) 原子がどれくらい強く相互作用するか(吸着エネルギー)」 を計算してみましょう。
通常、このような計算には「エンジン」として量子化学計算アプリ(VASP など)が必要ですが、ここでは ASE に内蔵されている EMT (Effective Medium Theory) という簡易的なモデルを使って、手軽にシミュレーションの流れを体験しましょう。
1. 表面と吸着原子の準備
まず、銅の (111) 面を作成し、そこに金原子を乗せる準備をします。
import matplotlib.pyplot as plt
from ase.build import fcc111, add_adsorbate
from ase.visualize.plot import plot_atoms
# 1. 銅 (Cu) の (111) 表面を作成
# size=(2, 2, 3) は x, y方向に2倍、z方向(厚さ)に3層という意味
# vacuum=10.0 は表面の上に10Åの真空層を作るという意味
slab = fcc111('Cu', size=(2, 2, 3), vacuum=10.0)
# 2. 金 (Au) 原子を吸着させる
# height=1.5 Å の高さに、'fcc' サイト(原子の隙間)に配置
add_adsorbate(slab, 'Au', height=1.5, position='fcc')
# 構造の確認(可視化)
fig, ax = plt.subplots(1, 2, figsize=(10, 5))
plot_atoms(slab, ax[0], radii=0.8, rotation=('0x,0y,0z')) # 上から
plot_atoms(slab, ax[1], radii=0.8, rotation=('-90x,0y,0z')) # 横から
ax[0].set_title("Top View")
ax[1].set_title("Side View")
plt.show()
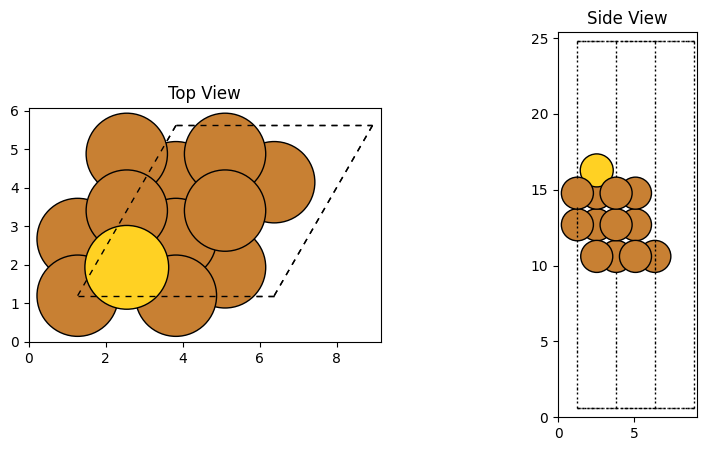
出力結果
2. 構造最適化とエネルギー計算
作成した構造は、原子を適当な位置に置いただけなので、エネルギー的に不安定なはずです。
構造最適化を行って、原子を最も安定な位置に動かして、そのエネルギーを計算しましょう。
吸着エネルギー
-
E_{slab+Au} -
E_{slab} -
E_{Au}
from ase.calculators.emt import EMT
from ase.optimize import BFGS
# --- 関数の定義: 構造最適化を行ってエネルギーを返す ---
def calculate_energy(atoms, label):
# 計算機 (Calculator) をセット。今回は軽量な EMT を使用。
atoms.calc = EMT()
print(f"--- {label} の構造最適化 ---")
# 構造最適化のアルゴリズム (BFGS) を設定
opt = BFGS(atoms)
# 構造最適化を実行 (力が 0.05 eV/Å 以下になるまで)
opt.run(fmax=0.05)
# ポテンシャルエネルギーを取得
energy = atoms.get_potential_energy()
print(f"{label} のエネルギー: {energy:.3f} eV\n")
return energy
# 1. 結合系 (Slab + Au) のエネルギー計算
# 先ほど作った slab には既に Au が乗っています
e_combined = calculate_energy(slab, "Slab + Au")
# 2. スラブ単体 (Slab only) のエネルギー計算
# 新しく Cu スラブを作り直して計算
slab_clean = fcc111('Cu', size=(2, 2, 3), vacuum=10.0)
e_slab = calculate_energy(slab_clean, "Clean Slab")
# 3. 吸着原子単体 (Au atom) のエネルギー計算
from ase import Atoms
atom_au = Atoms('Au', positions=[(0, 0, 0)])
# 単原子なので構造最適化は不要(動く余地がないため)ですが、calculatorをセットしてエネルギーを計算します
atom_au.calc = EMT()
e_au = atom_au.get_potential_energy()
print(f"Au atom のエネルギー: {e_au:.3f} eV\n")
# 4. 吸着エネルギーの算出
e_ads = e_combined - e_slab - e_au
print(f"=== 結果 ===")
print(f"吸着エネルギー: {e_ads:.3f} eV")
--- Slab + Au の構造最適化 ---
Step Time Energy fmax
BFGS: 0 11:21:30 5.783169 12.673172
BFGS: 1 11:21:30 3.681907 2.902334
BFGS: 2 11:21:30 3.532203 1.578831
BFGS: 3 11:21:30 3.437030 0.531280
BFGS: 4 11:21:30 3.407183 0.495493
BFGS: 5 11:21:30 3.390055 0.349153
BFGS: 6 11:21:30 3.381106 0.174141
BFGS: 7 11:21:30 3.377552 0.125389
BFGS: 8 11:21:30 3.376424 0.100673
BFGS: 9 11:21:30 3.375066 0.142941
BFGS: 10 11:21:30 3.373721 0.122262
BFGS: 11 11:21:30 3.373050 0.053427
BFGS: 12 11:21:30 3.372851 0.041777
Slab + Au のエネルギー: 3.373 eV
--- Clean Slab の構造最適化 ---
Step Time Energy fmax
BFGS: 0 11:21:30 2.853717 0.119250
BFGS: 1 11:21:30 2.852139 0.112307
BFGS: 2 11:21:30 2.840301 0.007200
Clean Slab のエネルギー: 2.840 eV
Au atom のエネルギー: 3.800 eV
=== 結果 ===
吸着エネルギー: -3.267 eV
計算された吸着エネルギーが 負の値 になっていれば、金原子は銅表面に吸着することでエネルギーが下がり、安定化している(くっつきやすい)ことを意味します。
このように、ASEを使えば「原子を配置する → 安定構造を探す → エネルギー差を計算する」というシミュレーションの基本的な手順を数行のコードで実装できます。
まとめ
今回は ASE を紹介しました。
- Point 1: Python で原子・分子シミュレーションを行うためのデファクトスタンダードです。
- Point 2: 30種類以上の外部計算エンジンを統一的に扱えるため、ツールの切り替えが簡単です。
- Point 3: 結晶や表面の作成機能がパワフルなので、構造データの準備だけでも利用価値があるはず。
本格的な解析では Gaussian などの外部計算エンジンと組み合わせて使いますが、まずは ASE 単体で動かしてみるだけでも、分子シミュレーションの面白さを体験できますよ。
ぜひ試してみてください。
Discussion