AlphaFold3の中身の日本語解説
AlphaFold3の中身の日本語解説
2024年5月21日(2024年6月13日更新)
佐藤 悠輝、古井 海里、大上 雅史(東京工業大学 情報理工学院)
⚠ 本記事で引用しているAlphaFold3論文[1]の図および表は、Springer Natureより著者らにpersonal, non-exclusive, non-transferable, non-sublicensable, revocable, world-wide Licenseが付与されています。これらの図および表の二次利用を禁止します。
(2024年6月13日更新) AlphaFold3論文[1:1]がCC BY 4.0ライセンスでオープンアクセスとして正式に出版されたことに伴いまして、同論文の図表等の二次利用はクリエイティブ・コモンズ・ライセンスに従って自由にできるようになりました。
1. はじめに
AlphaFold3[1:2]は、2024年にGoogle DeepMindとIsomorphic Labsによって共同開発された最新のタンパク質構造予測モデルです。2021年に発表された前バージョンに相当するAlphaFold2[2]ではアミノ酸配列からタンパク質の立体構造を予測する問題を扱っていましたが、AlphaFold3ではタンパク質、DNAやRNAなどの核酸、低分子リガンドを含む、様々な複合体の構造を高い精度で予測することができるようになりました。AlphaFold3の論文は速報版がNatureから日本時間2024年5月9日に出版されており、2024年5月21日現在オープンアクセスで読むことができます。
AlphaFold3は、従来のAlphaFold2やAlphaFold-Multimer[3]で用いられていた深層学習アーキテクチャと異なり、全原子の絶対座標を出力する拡散モデルをベースとしています。なお、本記事ではAlphaFold-Multimerも含めた前バージョンを「AlphaFold2」と呼ぶことにします。
アーキテクチャおよび学習方法の変更によって、AlphaFold3では新たに以下の立体構造予測が可能になりました。
- タンパク質-核酸複合体の立体構造予測
- タンパク質-低分子リガンド複合体の立体構造予測
- 共有結合による修飾や糖鎖修飾、非標準アミノ酸などを含むタンパク質の立体構造予測
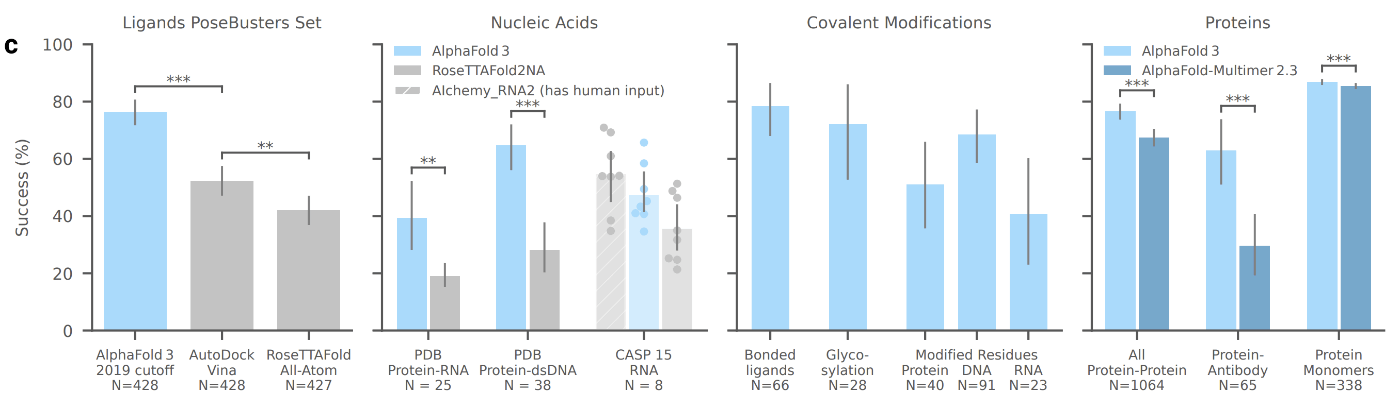
(AlphaFold3論文[1:3] Figure 1より引用, CC BY 4.0)
AlphaFold3の論文[1:4]では、以下のような主張が述べられています。
- タンパク質-低分子リガンド複合体の予測では、タンパク質の配列とリガンド構造式の情報のみから、PoseBusters[4]ベンチマークを用いて評価を行い、通常の(立体構造情報を使う)ドッキングシミュレーションよりも高い予測成功率を示しました。
- 核酸構造予測では、人手の評価が入った方法(Alchemy_RNA2)には劣ったものの、RoseTTAFoldNA[5]の予測精度を大きく上回りました。
- タンパク質や抗体の複合体予測の成功率が改善し、特に抗原抗体複合体の予測精度が向上しました。
本記事では、AlphaFold2やAlphaFold-Multimerからの差分に着目して、AlphaFold3のアーキテクチャや学習プロセスの全体を解説します。
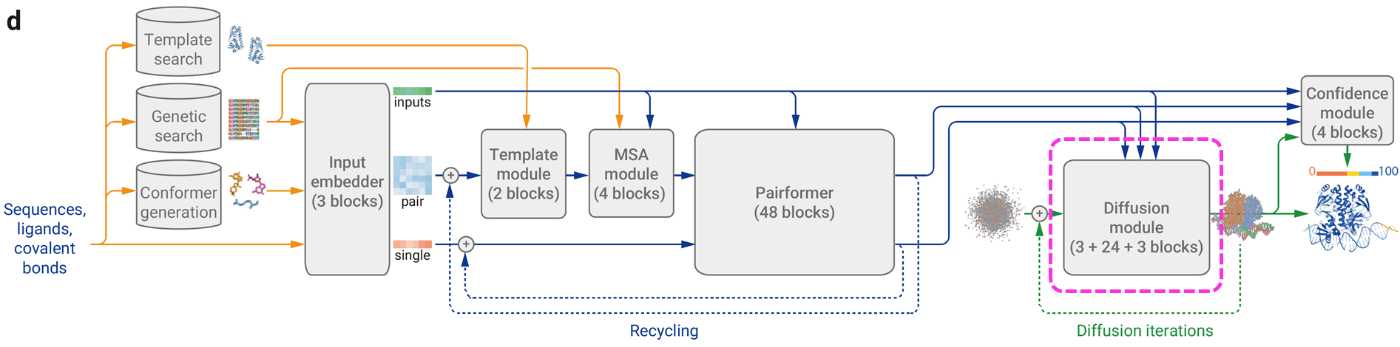
2. AlphaFold3の概要
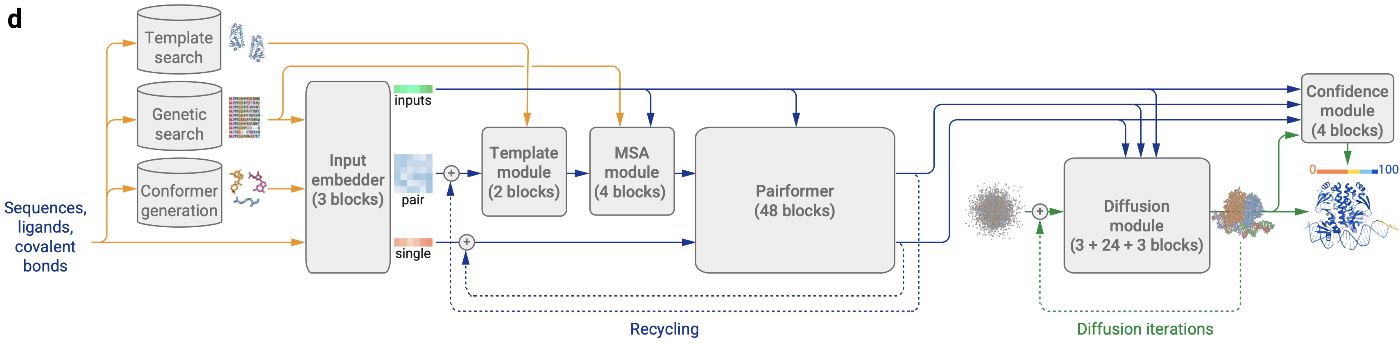
(AlphaFold3論文[1:5] Figure 1より引用, CC BY 4.0)
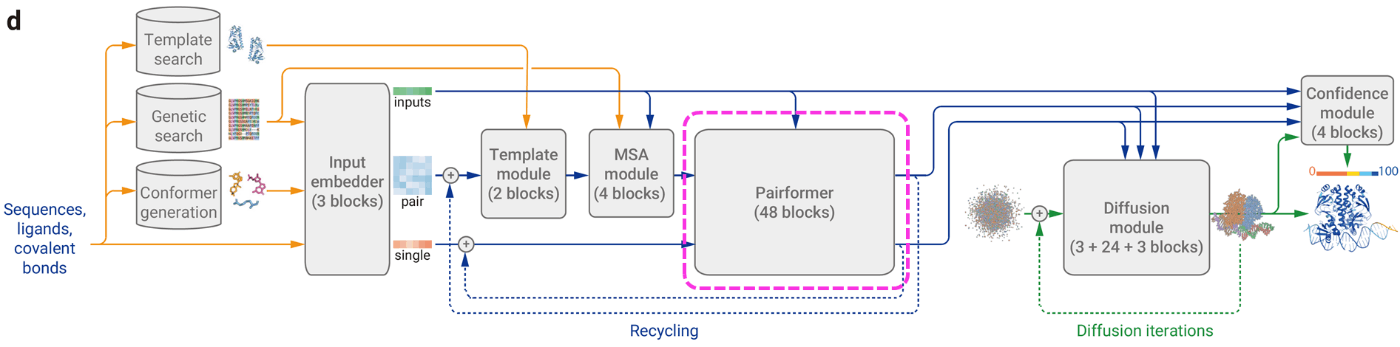
AlphaFold3での立体構造の推論は、入力データの前処理とトークン化とペア表現生成(MSAとテンプレート情報の導入)、Pairformerによる特徴量化、拡散モデルによる構造生成という流れで行われます。
モデルの構成の大枠はAlphaFold2がもとになっていますが、タンパク質以外への対応やタンパク質構造予測精度を向上させるためにいくつか変更が加えられています。AlphaFold2からの主な変更やAlphaFold3で新規な点は主に以下の通りです。
- 核酸やリガンドの入力に対応し、また残基の修飾にも対応。計算効率化のためにアミノ酸1残基や核酸1塩基に対しては1トークン、その他の化合物ではトークンは重原子(水素H以外の原子)1つに対して1トークン割り当てている。
- AlphaFold2のEvoformerを簡略化した「Pairformer」が導入され、多重配列アラインメント (MSA) がコアモジュールから削除された。
- AlphaFold3では拡散モデルによって構造を生成する。近年の構造生成などで一般的に扱われるSE(3)同変性(並進・回転させても同じ結果が得られる性質)を考慮していない。
- AlphaFold2では残基単位の移動ベクトルと側鎖のねじれ角を出力して全原子の座標を推定していたが、この方法はタンパク質以外への適用が難しかった。AlphaFold3では全原子の絶対座標を推論することでタンパク質以外の化合物の学習もできるようにした。
- 損失関数の変更。Mean Squared Error (MSE) lossをベースとしており、結合距離以外には立体違反や衝突を考慮していない。
- 4段階での学習。拡散モデルの学習を改善するためにAlphaFold2やAlphaFold3自身の予測構造を用いたデータセットを用いた学習(蒸留)を行っている。
全体としてアーキテクチャは洗練され、AlphaFold2よりもシンプルになった印象があります。以下ではAlphaFold3のアーキテクチャを順を追って見ていきたいと思います。
3. 入力情報のEmbedding
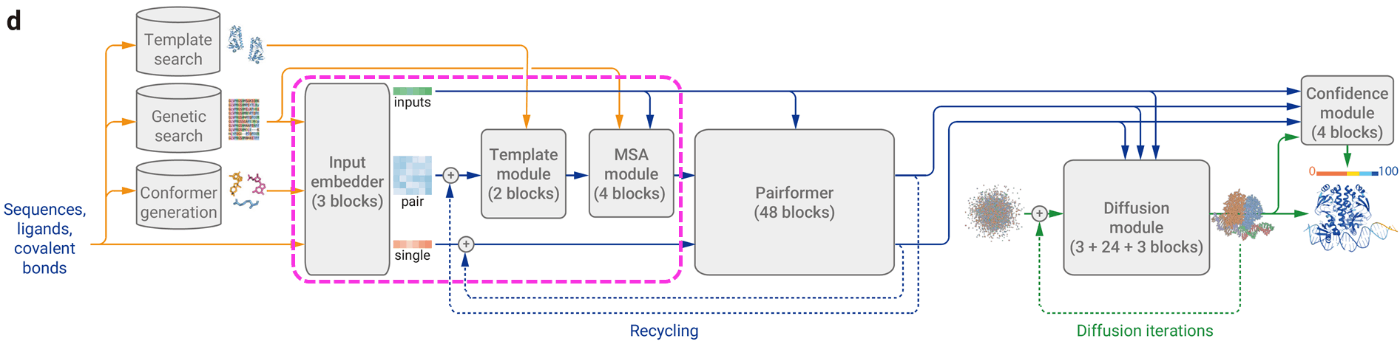
(AlphaFold3論文[1:6] Figure 1より引用, 追記, CC BY 4.0)
Input embedderブロック
最初の埋込表現は、残基の種類、参照配座、MSA特徴を単にconcatします。
- リガンドがCCD codeやSMILESによって与えられた際には、RDKitのETKDGv3を使用して参照配座を生成します。参照配座の特徴は、AtomAttentionEncoderによってpermutation invariantな方法で埋め込まれます。(リガンドの各原子をトークン配列として扱う際、これらのトークンの順序関係の違いが影響しないようにします。)
- MSA特徴は、Genetic search(配列データベース検索)の結果から得られた残基の欠損とMSAプロファイルから得られます。対象のデータベースと検索ツールは以下の表(タンパク質配列:Table 1、RNA配列:Table 2)に示されています。
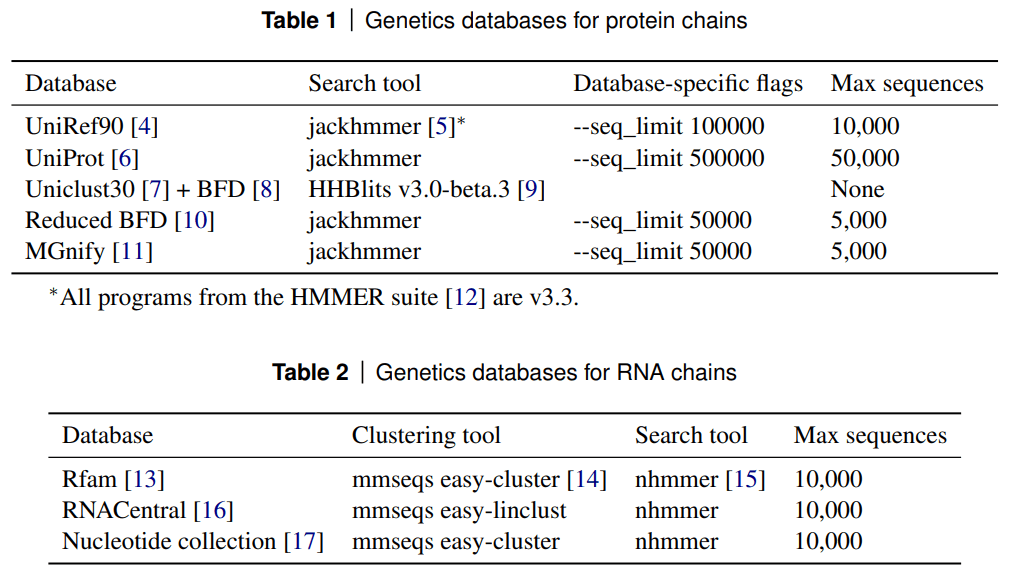 (AlphaFold3論文[1:7] Supplementary Tables 1, 2より引用, CC BY 4.0)
(AlphaFold3論文[1:7] Supplementary Tables 1, 2より引用, CC BY 4.0)
なお、入力特徴の細かい変更として、AlphaFold2のaatypeはアミノ酸20種+unknownの21次元で表していましたが、AlphaFold3のrestypeはアミノ酸21次元に加えて、DNAとRNAがそれぞれ4+1次元、gapが1次元、合計32次元に増えています(リガンドはunknown amino acidとして扱われます)。
また各トークンに対して、is_protein / rna / dna / ligandという4種類のマスクを持っており、これによって条件分けを行います。
Relative position encoding
- AlphaFold2から大きな変化はありませんが、AlphaFold3では原子単位でトークナイズする関係で、同一残基内のトークンについて相対的なトークンエンコーディングが行われます。
- entity, chain, residueそれぞれについて等しいかどうかで場合分けし、処理を変えて計算します。計算結果はone hot化してconcatし、線形変換してペア表現として出力されます。
Sequence-local atom attention (AtomTransformer)
原子レベルの情報を学習するため、配列の近傍の他の原子と「対話」するために導入されています。
- 原子配列を1次元配列に変換して各要素のほか要素に対するattentionを計算します。
- 局所的な原子配置の規則性を学習させる目的があるらしいです。
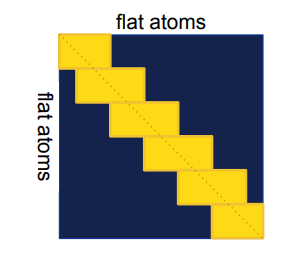
(AlphaFold3論文[1:8] Supplementary Figure 1より引用, CC BY 4.0)
この図のように、黄色の長方形の部分内でattentionが計算されることで局所的な規則性を学習します。
AtomAttentionEncoder
AtomTransformerを用いて、原子情報および座標情報をトークン表現にエンコードします。入力特徴量を作る際は参照コンフォーマーが座標情報として与えられます。また、Diffusion moduleでも用いられます。
- 各原子の位置、元素種、電荷などの特徴量から単一の原子表現を作成
- 原子対間のペア表現の作成
- 既存のトークン表現があれば、それを組み込む(Diffusion module用)
- AtomTransformerにより、近傍の原子情報を用いて更新
AtomAttentionDecoder
AtomAttentionDecoderはDiffusionModule内でのみ用いられます。トークン表現を原子表現に変換した後AtomTransformerで更新し、原子座標の更新量を予測します。
MSAとテンプレート情報の導入 (MSA module)
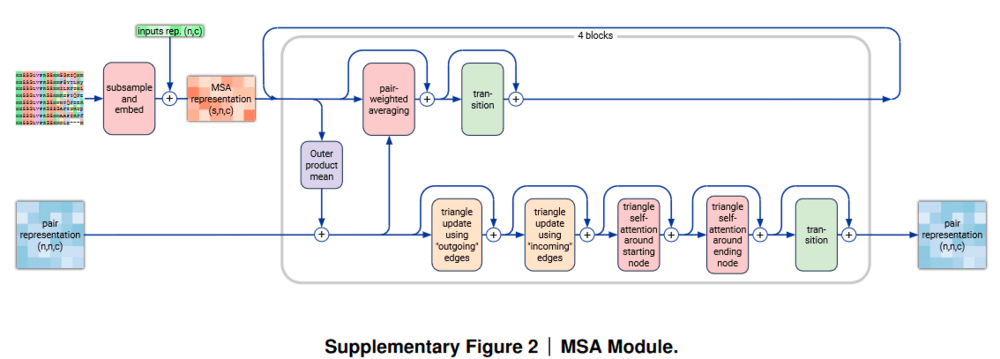 (AlphaFold3論文[1:9] Supplementary Figure 2より引用, CC BY 4.0)
(AlphaFold3論文[1:9] Supplementary Figure 2より引用, CC BY 4.0)
MSA配列はAlphaFold-multimerと同様の方法で用意されます。MSA moduleではPair representation(ペア表現)とMSA representationを入力として4ブロックで構成されます。
AlphaFold2と異なり、MSAに対して用いられていたaxial self-attentionがなくなっています。すなわち、MSAに対して行をまたいだattention処理は行われません。AlphaFold3のでは、タンパク質や核酸に関するできるだけ多くの情報を含むべきという動機で全ての情報をペア表現に集約したと主張しています。
なお、AlphaFold2のEvoformerでMSAに対して行った処理に類似しており、ペア表現のTriangle UpdatesについてはAlphaFold2から変更はなさそうです。また、MSA moduleのあとにテンプレート特徴が作成されたペア表現が導入されます。AlphaFold2のtemplate_pair_featに相当する特徴量のみが追加されています。
ペア表現にテンプレート情報とMSA情報をそれぞれ追加した後、ペア表現はPairformerの入力として渡されます。
4. Pairformer
 (AlphaFold3論文[1:10] Figure 1より引用, 追記, CC BY 4.0)
(AlphaFold3論文[1:10] Figure 1より引用, 追記, CC BY 4.0)
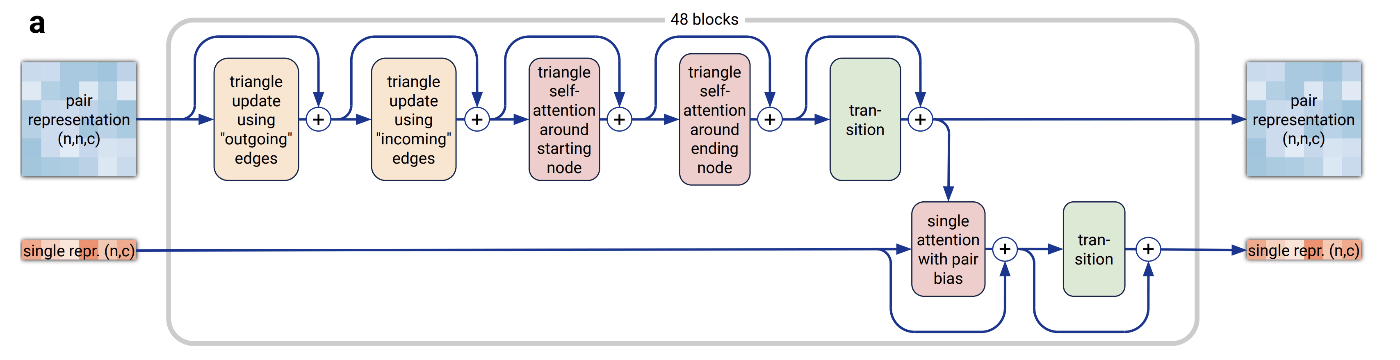 Pairformerの構造(AlphaFold3論文[1:11] Figure 2aより引用, CC BY 4.0)
Pairformerの構造(AlphaFold3論文[1:11] Figure 2aより引用, CC BY 4.0)
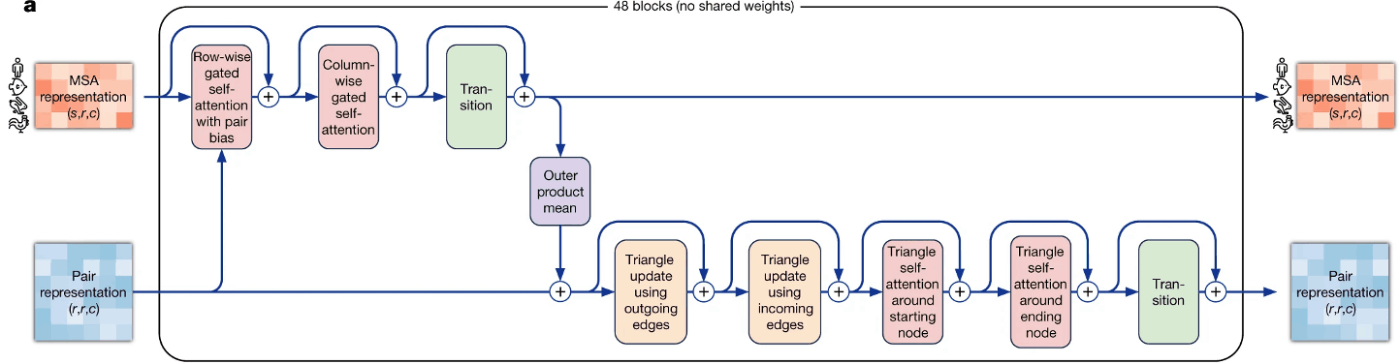 Evoformerの構造(AlphaFold2論文[2:1] Figure 3aより引用, CC BY 4.0)
Evoformerの構造(AlphaFold2論文[2:1] Figure 3aより引用, CC BY 4.0)
AlphaFold3のPairformerはAlphaFold2のEvoformerとよく似ています。PairfomerではMSAを入力に取らないため、MSAに関係する処理が削除されている点がEvoformerと異なります。Evoformerと同様に48ブロックを反復的に適用します。
なお、AlphaFold2のTransitionブロックではReLUが用いられていましたが、AlphaFold3ではSwiGLU[6]に変更されています。
5. Diffusion module -- 拡散モデルによる構造生成
 (AlphaFold3論文[1:12] Figure 1より引用, 追記, CC BY 4.0)
(AlphaFold3論文[1:12] Figure 1より引用, 追記, CC BY 4.0)
拡散モデルについて
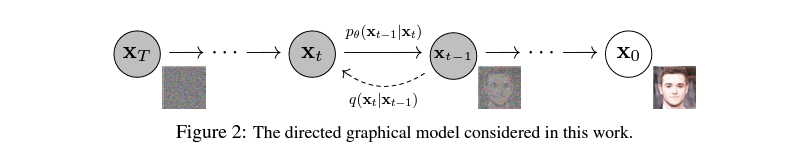 (DDPM論文[7] Figure 2より引用)
(DDPM論文[7] Figure 2より引用)
拡散モデルは生成モデルの1つです。拡散モデルでは、ラベルデータ
拡散過程では一般的にラベルデータにガウスノイズをステップ数だけ付与します。逆拡散過程では、拡散過程の各ステップにおけるノイズ付与後のデータとステップ数を用いてノイズ付与前のデータを推論するように学習が行われます。そのため、逆拡散過程の最後のステップではノイズ付与前のラベルデータを推論します。
拡散モデルは複雑な生成過程を複数ステップに分解し各ステップを独立して学習可能であり、損失関数もラベルデータとの平均2乗誤差というシンプルな関数であるため安定して複雑なデータ分布を学習可能な点が強みです。さらに、適切なモデルを利用して逆拡散過程の各ステップに任意のデータを条件付けしてほしい出力を得ることができます。拡散モデルは他の生成モデルと比較して、条件を反映したデータ生成の精度が高い場合が多いです[8]。
Diffusion moduleの全体像
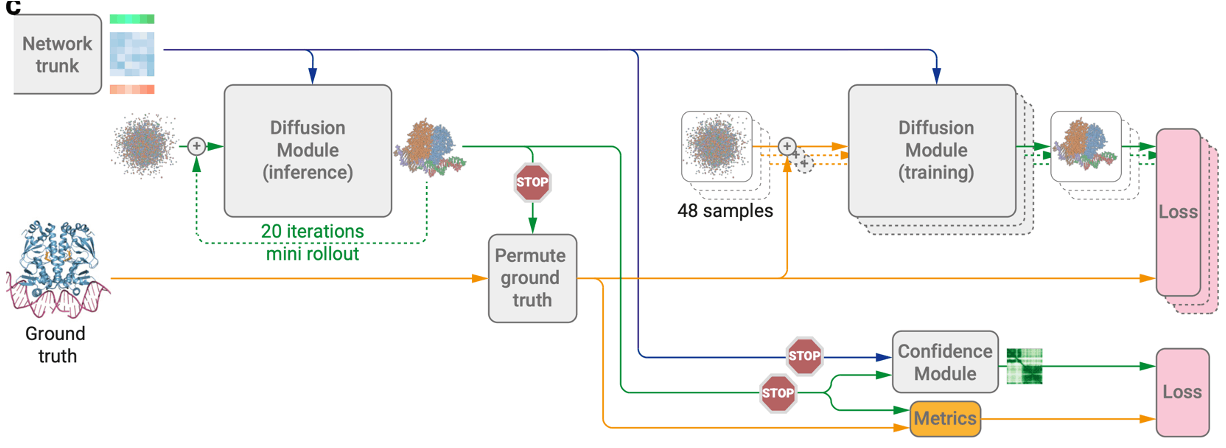 (AlphaFold3論文[1:13] Figure 2cより引用, CC BY 4.0)
(AlphaFold3論文[1:13] Figure 2cより引用, CC BY 4.0)
AlphaFold3では、AlphaFold2と異なり、拡散モデルを用いて全原子の絶対座標を推論します。上の図において、青の矢印は条件付けに使用するデータ、緑の矢印は推論データ、黄色の矢印: 正解データ、stop記号はその先の計算について勾配計算を行わない(勾配停止)ことを示しています。
AlphaFold3では拡散モデルのアーキテクチャ、学習方法について大部分はEDM[9]に従っています。EDMはサンプリング手法・学習プロセスについて改善することで、生成データの品質を維持したまま推論時のステップ数を大きく削減した手法です。
AlphaFold3ではAlphaFold2と異なり全原子の絶対座標を推論しておりSE(3)同変性を考慮する学習がなされていません。これについて論文内でも言及があり、“現在の傾向とは対照的である”としつつも、論文を1本引用[10]した上でSE(3)同変性を考慮しなくても問題ないと述べています。
入力ノイズ (48 samples) の作成
正解構造データに対してノイズ付与を行います。この時、Mini rolloutにより対応付けが行われた正解構造データを使用します。
また、学習効率向上のため1つのデータに対して48の独立したノイズデータを作成します。作成手順は以下の通りです。
- 正解構造データに対してランダムな回転・並進を適用
- ノイズを付与(ノイズのサンプリング方法はEDMに従う)
逆拡散過程 (Diffusion Module)
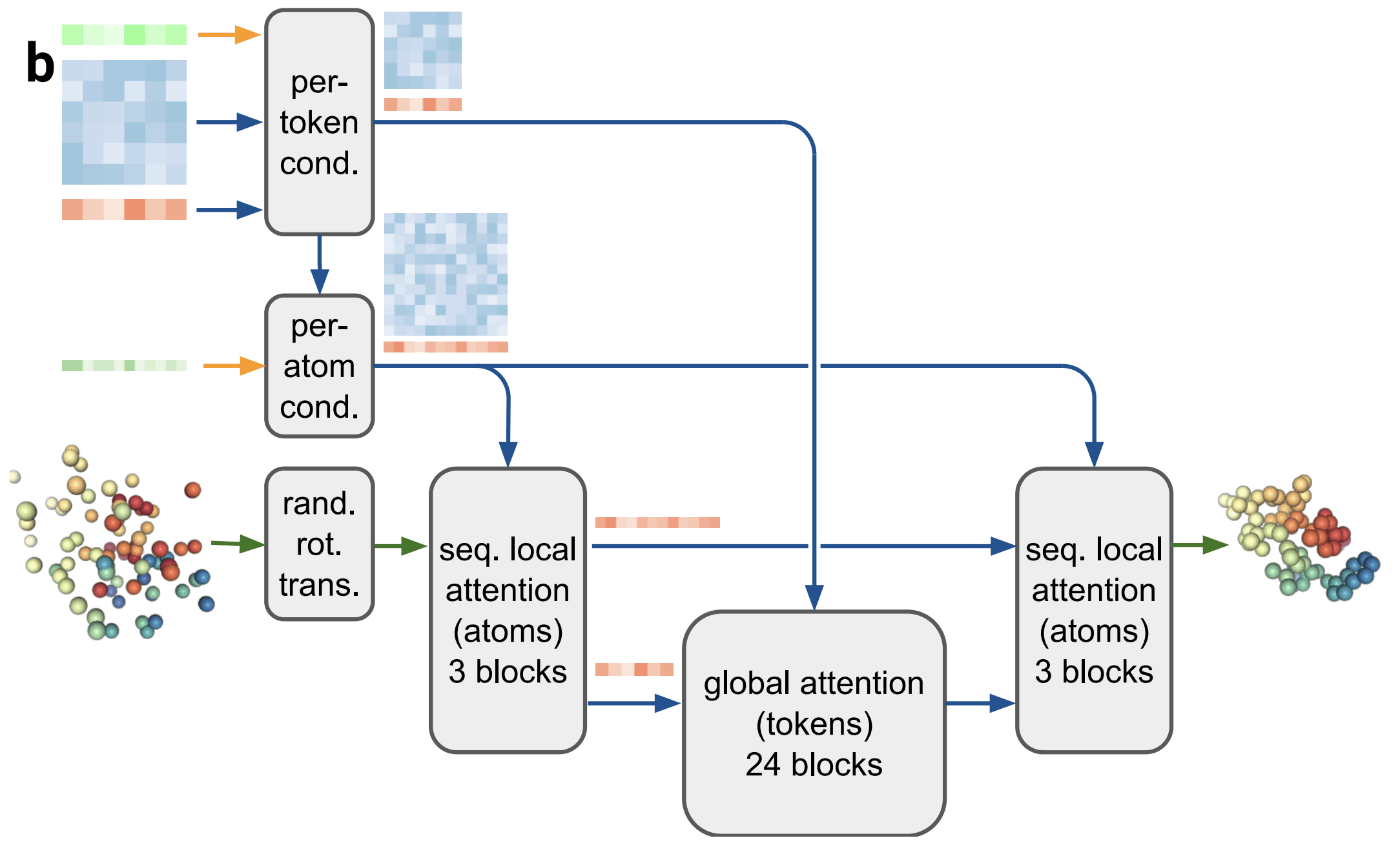 (AlphaFold3論文[1:14] Figure 2bより引用, CC BY 4.0)
(AlphaFold3論文[1:14] Figure 2bより引用, CC BY 4.0)
逆拡散過程の各ステップで以下の4つのデータを条件付けしています。
- 原子単位の埋め込み前情報(上から4番目の緑の配列)
- トークン単位の入力埋め込み表現(上から1番目の黄緑の配列)
- Pairformerの出力(ペア表現:上から2番目の青の行列、シングル表現:上から3番目の赤の配列)
拡散モデルの各逆拡散過程の処理手順は以下の通りです。
- (rand. rot. trans.) 1ステップ前の推論構造に対してランダムな回転・並進移動を行います。並進は1 Å以内と考えられ世界座標系の中心から大きく離れないようなデータ拡張と考えられます。以後これを入力構造と呼びます。
- (per token cond.) トークン単位のペア表現・シングル表現に対して条件付けを行います。ペア表現に対しては相対位置情報を、シングル表現に対しては入力埋め込み表現とステップ数をconcatして線形変換により特徴量を獲得します。
- (seq. local attention) 原子単位の埋め込み前情報を用いてバイアスアテンションを計算することで原子間の局所的な構造情報に関する特徴を抽出します。最後に得られた原子単位の特徴量をトークン単位の特徴量に変換します。
バイアスアテンションとは、AlphaFold2でMSAに対して使用されたMSA row-wise gated self-attention with pair biasと似た仕組みです。下図のようにMSAではなく入力特徴量を用いて計算されます。ペア表現には原子間の相対位置など原子同士の関連性に関する情報が埋め込まれており、これをバイアスとして加算することで関連度が高い原子間の重みを大きくすることができます。
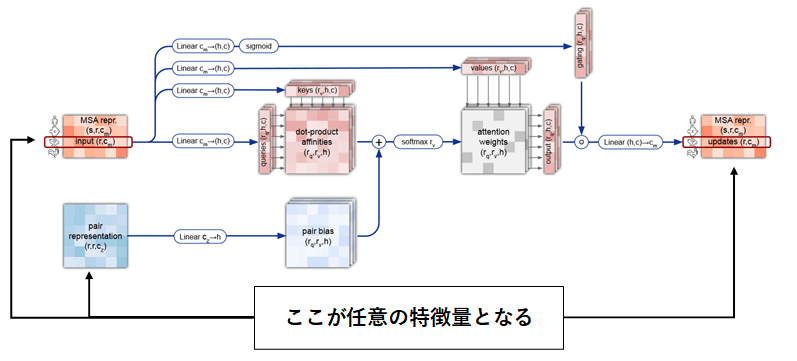
(AlphaFold2論文[2:2] Supplementary Figure 2 に変更点を追記, CC BY 4.0) - (global attention) 3.で得られたトークン単位の特徴量を用いてバイアスアテンションを計算することでトークン間の大域的な特徴を抽出します。ここでバイアスとして用いるペア表現には2.で条件付けされたトークン単位のペア表現が用いられます。
- (seq. local attention) 4.で得られたトークン単位の特徴量を原子単位の特徴に変換し、これを用いてバイアスアテンションを計算し構造更新に必要な原子単位の情報を生成します。この時用いられるバイアスには3.と同じものが使用されます。
- ノイズ付与した初期構造と5.の出力を用いてデノイズ後の構造を生成します。
6. 信頼度の予測
Mini rollout
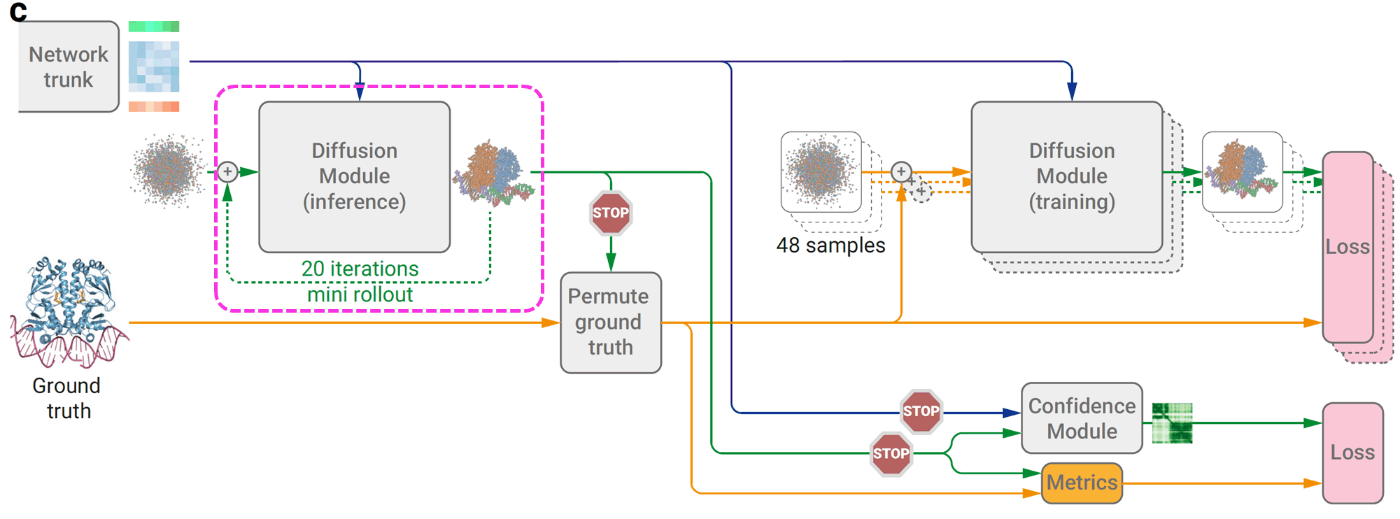
(AlphaFold3論文[1:15] Figure 2cより引用, 追記, CC BY 4.0)
Confidence moduleのいくつかのヘッドで予測構造が必要になるため、勾配を計算しない状態でランダムなノイズから20 step推論して出力を得ます。
なお、同じ配列を持つタンパク質や核酸やリガンドが含まれる場合、正解構造と同じ順番でチェインを予測するとは限りません。そこで、AlphaFold-Multimerで取られていたようにMulti-Chain Permutation Alignmentを実施することで、正解構造と予測構造の対応付けを行っています。
Confidence head
AlphaFold3ではpLDDT (predicted local distance difference)、pAE (pairwise atom-atom aligned error) に加えてpDE(予測距離誤差)の3つの信頼度指標と、実験的に解かれた構造かどうかを予測します(pDEのみがAlphaFold3で新規)。なお、pLDDTやpAEの計算では、核酸やリガンドについても値を算出するための変更が加わっています。なお、これらはConfidence headで学習され、学習中の単一/ペア表現や予測構造に対しては勾配が停止されます。
- Predicted local distance difference test (pLDDT)
- pLDDTは原子とのしきい値内の距離にある原子ペアについてその距離が保存されているものの割合を意味するLDDTの予測
- AlphaFold2のpLDDTからの変更点
- リガンド原子の場合はリガンド内の原子は考慮されない。
- 距離のしきい値は、タンパク質原子との距離が15 Å未満、核酸原子との距離が30 Å未満
- アミノ酸の場合はCα原子、核酸の場合はC1'原子の座標で距離を計算する。
- Predicted aligned error (pAE)
- pAEはトークンのペア間での相対的な位置関係のずれを予測する。
- pAEでは、この相対的な位置関係のずれを計算するために、各トークンの"座標系"を3つの原子
(a_i, b_i, c_i) - AlphaFold2のpAEからの変更点として、アミノ酸では
(N, C^\alpha, C) (C1', C3', C4') b_i a_i c_i
- Predicted distance error (pDE)
- 原子間の絶対距離の誤差を直接予測した値(AlphaFold3で新規)。
7. 損失関数
全体の損失関数
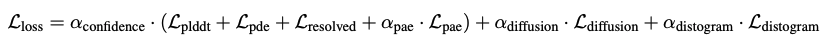 (AlphaFold3論文[1:16] 式(15)より引用, CC BY 4.0)
(AlphaFold3論文[1:16] 式(15)より引用, CC BY 4.0)
AlphaFold3の場合は以下の5つで表されます。
-
\mathcal L_{\rm plddt} \mathcal L_{\rm pde} \mathcal L_{\rm pae} \mathcal L_{\rm pae} -
\mathcal L_{\rm diffusion} -
\mathcal L_{\rm distogram}
また、AlphaFold2では構造的な違反に関する損失
Diffusion moduleの損失
Diffusion moduleの損失は以下の式で表されます。
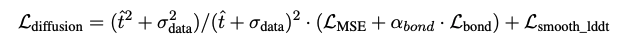 (AlphaFold3論文[1:17] 式(6)より引用, CC BY 4.0)
(AlphaFold3論文[1:17] 式(6)より引用, CC BY 4.0)
基本的には以下の損失の組み合わせです。
-
\mathcal L_{\rm MSE} -
\mathcal L_{\rm bond} -
\mathcal L_{\rm smooth\_lddt}
また、第一項の係数は拡散プロセスにおけるノイズレベルに基づいて決められる重みで、ノイズレベルが高いほどこの項が大きくなります。この損失関数はベースとなっている拡散モデルであるEDMで提案されています。
8. AlphaFold3の学習と推論
データセット
 (AlphaFold3論文[1:18] Supplementary Table 3より引用, CC BY 4.0)
(AlphaFold3論文[1:18] Supplementary Table 3より引用, CC BY 4.0)
PDB構造以外に、4つの蒸留 (distillation) データセット、転写因子に関するデータセットを用います。上表から分かるように、PDBデータセットとmonomerの蒸留へのweightがほとんどを占めています。
PDBデータセット
PDBデータについては、種類
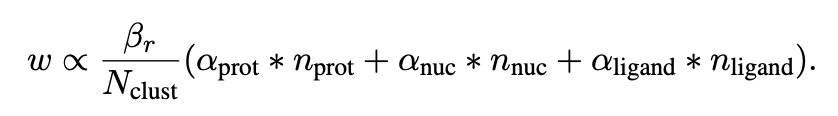 (AlphaFold3論文[1:19] 式(1)より引用, CC BY 4.0)
(AlphaFold3論文[1:19] 式(1)より引用, CC BY 4.0)
ここで、
これらのchain/interfaceを中心に、学習時の計算リソースの節約かつ相互作用interfaceの効率的な学習を目的として、配列のcroppingを行います。croppingはAlphaFold-Multimerと同様、contiguous cropping(参照原子と連続した原子を削除)、spatial cropping(参照原子と空間的に近い原子を削除)、spatial interface cropping(参照となる界面原子と空間的に近い原子を削除)の3種が行われます。
モデルの訓練では、データのバイアスを減らすために、chainおよびinterfaceでのクラスタリングを行って冗長性を減らしています。タンパク質は40%配列相同性
※注)「配列相同性」は原文ではn% sequence homologyという言葉遣いがされており、n% sequence identityとは異なるものです。すなわち、「100% sequence homology」が配列の完全一致を意味しない可能性があります。このあたり、論文中では曖昧な表現がなされており、詳細がわかりません。
蒸留 (distillation) データセット
PDBデータセットのGround Truthの存在する立体構造以外にも、4つのデータセットがAlphaFold2/3予測構造から蒸留データセットとして取得されました。このうち3つは従来のAlphaFold2から、RNA蒸留データセットと転写因子データセットはAlphaFold3の予測から得られています。
このうち、disorder領域のPDB蒸留は、AlphaFold3でdisorder領域に嘘の二次構造(幻覚, hallucination)が発生してしまうことを防ぐ目的で導入されました。
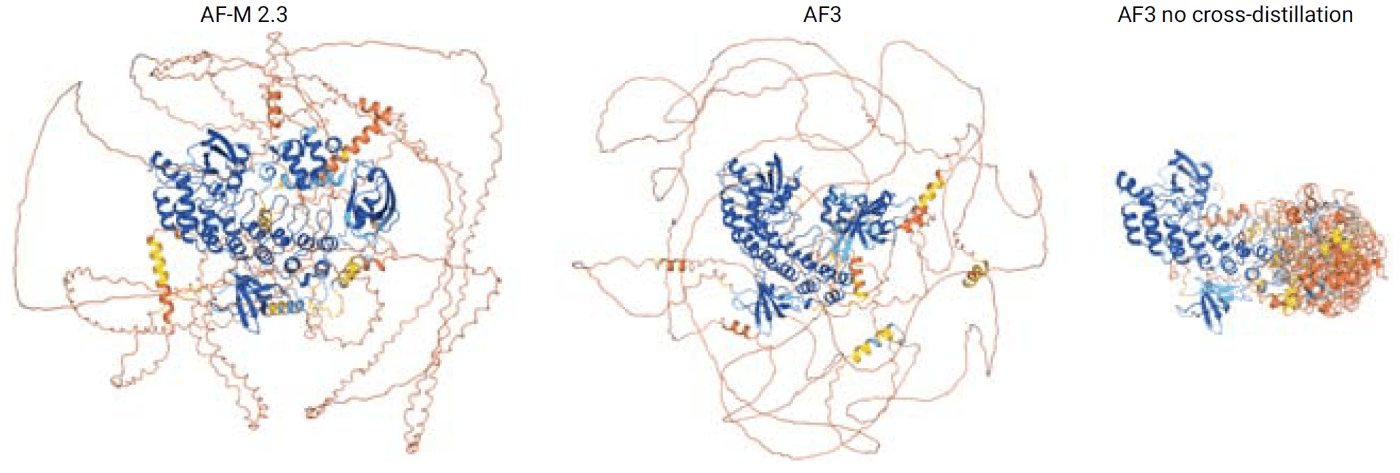
(AlphaFold3論文[1:20] Extended Data Fig. 1より引用, CC BY 4.0)
AlphaFold2では、disorder領域では特有の"リボン状"の予測を返します(図の左)。disorder領域のPDB蒸留データセットを使わないで学習したAlphaFold3は、disorder領域で幻覚を起こします(図の右)。これはAlphaFold3で拡散モデルを使用したモデルに移行したためであり、幻覚は生成モデルで生じやすい問題として知られています[11]。AlphaFold3ではdisorder領域の挙動をAlphaFold2に近づけるために、disorder領域のPDB蒸留データセットを用いてAlphaFold2予測構造を学習に使っています(図の中央)。
- タンパク質単量体の蒸留
AlphaFold-Multimerと同様、MGnifyデータベースから200残基を超えるAlphaFold2予測構造 - 短いタンパク質単量体の蒸留
4〜200残基のMGnify配列に対するAlphaFold2予測構造(単量体の蒸留と同様)。単量体の上流データセットは合わせて約4100万構造ある。 - タンパク質disorder領域のPDB蒸留
PDB構造に対するAlphaFold-Multimerの予測構造を用いる。この際、disorder領域の40残基とAlphaFold予測構造に対してGDT(global distance test、予測構造と実験構造の間の類似性を評価する指標)が60以上となる衝突のない予測構造を25,000個収集した。この蒸留データセットは、diorder領域の予測がAlphaFold DBの予測と一致させるために用いられる。 - RNA蒸留
RfamをMMseqs2で90%配列同一性と80% coverageでクラスタリングし、クラスタごとにクラスタ代表を1本取得し、これらの配列のうち10残基以上かつAlphaFold3のpDEが2以下という条件に適合した65,000個のAlphaFold3構造を蒸留データセットとして使用した。 - 正例の転写因子蒸留
実際の転写因子プロファイルに基づき、低いpDEを持つAlphaFold3による転写因子-DNAペアの予測構造を蒸留データセットとして作成する。- JASPAR、SELEXデータセットから転写因子配列を取得し、10%配列相同性と60%のカバレッジでクラスタリングして半数のクラスタの転写因子配列を選択。
- 各転写因子に対し、JASPARの位置頻度行列(PFM)からランダムにDNAモチーフをサンプリングし、DNAモチーフ、転写因子配列、DNA相補鎖を組み合わせて正例を作成。
- AlphaFold3で予測構造を生成し、タンパク質-DNAペアのpDEが5 Å以下のものを使用し、1,165個のユニークなタンパク質配列を含む16,439個のタンパク質-DNA複合体蒸留データセットが作成された。
- なお、この蒸留データセットでは、訓練時にランダムにDNAを最大100塩基まで伸ばす処理が加えられている。伸ばす際には構造が理想的なB-DNAらせんになるように構築する。
- 負例の転写因子蒸留
- おそらく過学習(存在しない転写因子-DNA結合を予測)を防ぐために導入された。
- ランダムに60〜100塩基長のDNA配列とその相補鎖を生成。
- タンパク質モノマー蒸留セットからランダムに予測構造を組み合わせ、タンパク質とDNAを一定距離
(25 \mathrm{\ Å}+\mathcal N(2,3)\mathrm{\ Å})
学習ステージ
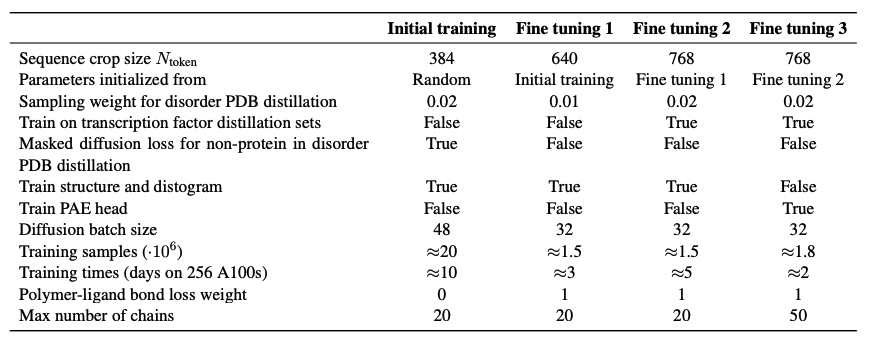 (AlphaFold3論文[1:21] Supplementary Table 6より引用, CC BY 4.0)
(AlphaFold3論文[1:21] Supplementary Table 6より引用, CC BY 4.0)
学習は初期学習、ファインチューニング1, 2, 3の4段階に分けて実施されます。Supplementary InformationのTableの内容は大まかに以下のようになっています。
- 学習時のトークンのcrop数を384、640、768、768と徐々に大きくする
- 最終ステップ(ファインチューニング3)でdistogramの学習からPAEヘッドの学習に切り替え
- ファインチューニング2から転写因子蒸留セットを学習
- 全体の学習は256個のA100で20日程度要する
- サンプル数やバッチサイズ、チェインの数、disorderPDB蒸留の重みなどは学習ステージごとに変更
また1ステップ目のみdisorder PDB distillationでタンパク質以外のdiffusion lossをマスクして学習しています。
推論時間

(AlphaFold3論文[1:22] Supplementary Table 8より引用, CC BY 4.0)
推論時間はトークン数(タンパク質:残基数、DNA/RNA:塩基数、リガンド:重原子数)が増えるごとに増大します。上記はA100 GPUを16枚使用したときの値です(recycles 10で実行。MSA構築や後処理などの時間は含まれていません)。
9. まとめ
本記事では、タンパク質、核酸、低分子、イオン、修飾残基などの複合体構造予測が大幅に改善されたAlphaFold3のアーキテクチャや学習プロセスについて紹介しました。記事執筆(2024/5/21)時点ではコードの実装は公開されておらず、機能が制限された alphafoldserver.com(ベータ版)のみが公開されているだけですが、詳細なアルゴリズムや学習手順は本記事で記載したように論文およびSupporting Informationに情報があります(再現実装もできる程度の情報があります)。推測に過ぎませんが、今後DeepMindが実装を公開したり、論文で言及されている全ての機能が利用可能なサービスを提供したりするかもしれません[12](ただしアカデミック利用に限定されそうです)。また、OpenFold[13]のように、実装から学習までの全てを再現するOSSプロジェクトもすでに発足されているようです[14]。
立体構造予測問題の解決が直ちに創薬の解決を意味するわけではありません[15]が、タンパク質と様々な分子の複合体が高精度に予測できることは、創薬の初期段階における分子設計の加速が期待できます。訓練データに含まれていないような分子間相互作用様式をどの程度まで予測できるか、アロステリックサイトや異なるエピトープへの結合を正しく予測できるか、結合しないはずの分子リガンドに対して嘘を出力しないのか、
-
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, Bodenstein SW, Evans DA, Hung CC, O'Neill M, Reiman D, Tunyasuvunakool K, Wu Z, Žemgulytė A, Arvaniti E, Beattie C, Bertolli O, Bridgland A, Cherepanov A, Congreve M, Cowen-Rivers AI, Cowie A, Figurnov M, Fuchs FB, Gladman H, Jain R, Khan YA, Low CMR, Perlin K, Potapenko A, Savy P, Singh S, Stecula A, Thillaisundaram A, Tong C, Yakneen S, Zhong ED, Zielinski M, Žídek A, Bapst V, Kohli P, Jaderberg M, Hassabis D, Jumper JM. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024. (2024/5/21時点, accepted manuscript) doi: 10.1038/s41586-024-07487-w ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎ ↩︎
-
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873): 583-589, 2021. doi: 10.1038/s41586-021-03819-2. ↩︎ ↩︎ ↩︎
-
Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D. Protein complex prediction with AlphaFold-Multimer. bioRxiv preprint, 2021.10.04.463034v2, 2021. doi: 10.1101/2021.10.04.463034 ↩︎
-
Buttenschoen M, Morris GM, Deane CM. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem Sci, 15(9): 3130-3139, 2024. doi: 10.1039/d3sc04185a ↩︎
-
Baek M, McHugh R, Anishchenko I, Jiang H, Baker D, DiMaio F. Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA. Nat Methods, 21(1): 117-121, 2024. doi: 10.1038/s41592-023-02086-5 ↩︎
-
Shazeer N. Glu variants improve transformer. arXiv preprint, arXiv:2002.05202, 2020. doi: 10.48550/arXiv.2002.05202 ↩︎
-
Ho J, Jain A, Abbeel P. Denoising diffusion probabilistic models. Advances in Neural Information Processing Systems (NeurIPS 2020), 33, 6840-6851, 2020. pdf ↩︎
-
Ho J, Salimans T. Classifier-free diffusion guidance. NeurIPS 2021 Workshop, arXiv:2207.12598, 2021. doi: 10.48550/arXiv.2207.12598 ↩︎
-
Karras T, Aittala M, Aila T, Laine S. Elucidating the Design Space of Diffusion-Based Generative Models. arXiv preprint, arXiv:2206.00364, 2022. doi:10.48550/arXiv.2206.00364 ↩︎
-
Wang Y, Elhag AA, Jaitly N, Susskind JM, Bautista MA. Swallowing the Bitter Pill: Simplified Scalable Conformer Generation. arXiv preprint (ICML2024 accepted), arXiv:2311.17932, 2023. doi:10.48550/arXiv.2311.17932 ↩︎
-
Ji Z, Lee N, Frieske R, Yu T, Su D, Xu Y, Ishii E, Bang YJ, Madotto A, Fung P. Survey of hallucination in natural language generation. ACM Computing Surveys, 55(12): 1-38, 2023. doi: 10.1145/3571730 ↩︎
-
Pushmeet Kohli @pushmeet, "We love the excitement & results from the community on AlphaFold 3 and are doubling the AF Server daily job limit to 20. Happy to also share that we're working on releasing the AF3 model (incl weights) for academic use, which doesn’t depend on our research infra, within 6 months." https://x.com/pushmeet/status/1790086453520691657 ↩︎
-
Ahdritz G, Bouatta N, Floristean C, Kadyan S, Xia Q, Gerecke W, O'Donnell TJ, Berenberg D, Fisk I, Zanichelli N, Zhang B, Nowaczynski A, Wang B, Stepniewska-Dziubinska MM, Zhang S, Ojewole A, Guney ME, Biderman S, Watkins AM, Ra S, Lorenzo PR, Nivon L, Weitzner B, Ban YA, Chen S, Zhang M, Li C, Song SL, He Y, Sorger PK, Mostaque E, Zhang Z, Bonneau R, AlQuraishi M. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat Methods, 2024. doi: 10.1038/s41592-024-02272-z ↩︎
-
kyegomez/AlphaFold3: Implementation of AlphaFold 3 from the paper: "Accurate structure prediction of biomolecular interactions with AlphaFold3" in PyTorch https://github.com/kyegomez/AlphaFold3 ↩︎
-
AlphaFold 3 Debuts | Science | AAAS https://www.science.org/content/blog-post/alphafold-3-debuts ↩︎
Discussion