Snowflakeを用いたVCFフォーマットの遺伝子バリアントデータと表現型データベースとの統合
データエンジニア兼バイオインフォマティシャンの是枝です。
業務でsnowflakeを触っていますが、snowflakeの性能は素晴らしいものがあります。一方で、私は生物系研究者でもあり、普段はバイオインフォマティクスで研究しています。ゲノムデータを扱った解析をコンピュータの力で解析することがメインですが、ふと、snowflakeを使えばもっといい解析ができるのでは...?と思うことが増えてきています。そこでsnowflakeでできるバイオインフォマティクスにチャレンジしてsnowflakeとバイオインフォマティクスのコラボレーションの可能性を探ってみたいと思います。前半はVCFフォーマットの遺伝子バリアントデータとアノテーションデータをsnowflake上で統合してみます。後半は、snowflakeでバイオインフォマティクスに関して個人的意見を述べます。
ハンズオンは基本的にはこちらのバリアントデータ解析の記事の内容に則って解析を進めます。Snowflakeで実際のゲノムデータセットを取り込み、クエリを実行するエンドツーエンドの例を説明しています。トライアルアカウントで1時間くらいで実行できました。
Snowflakeで公開ゲノムデータセットを扱う準備をする
何はともあれ、まずは解析するデータを用意する必要があります。illuminaがOpen Data Registryで利用可能な一般公開されているバリアントデータとアノテーションデータをAWS上にホストしてくれているみたいなのでこちらを使います。今回は1000-Genomesプロジェクトのシーケンスコレクションから、3,202人分のDRAGENを用意します。ちなみにDRAGENとはDynamic Read Analysis for Genomicsの略で、Illumina社が提供しているバイオインフォマティクスのための専門的なハードウェアおよびソフトウェアのプラットフォームを指します。DRAGENは高速で精度の高いゲノム配列情報の解析を行い、シーケンシングデータからバリアントを検出する能力を持っています。またこちらの配列はGRCh38というヒトのリファレンスゲノムにマッピングされた配列サンプルです。
Open Data RepositoryのS3と外部ステージを作成します。下記クエリの実行時間はxsサイズで4分59秒でした。
-- Data location from 1000-genomes files
-- Create stage including Directory Table
create or replace stage dragen_all
directory = (enable = true)
url = 's3://1000genomes-dragen-3.7.6/data/individuals/hg38-graph-based'
file_format = (type = CSV compression = AUTO)
;
VCF (Variant Call Format) ファイルが取得されています。VCFファイルとは、ゲノム配列のバリアント(遺伝的変異)を表すために用いられる標準的なファイル形式です。それぞれのVCFファイルには約500万行のデータが含まれています。
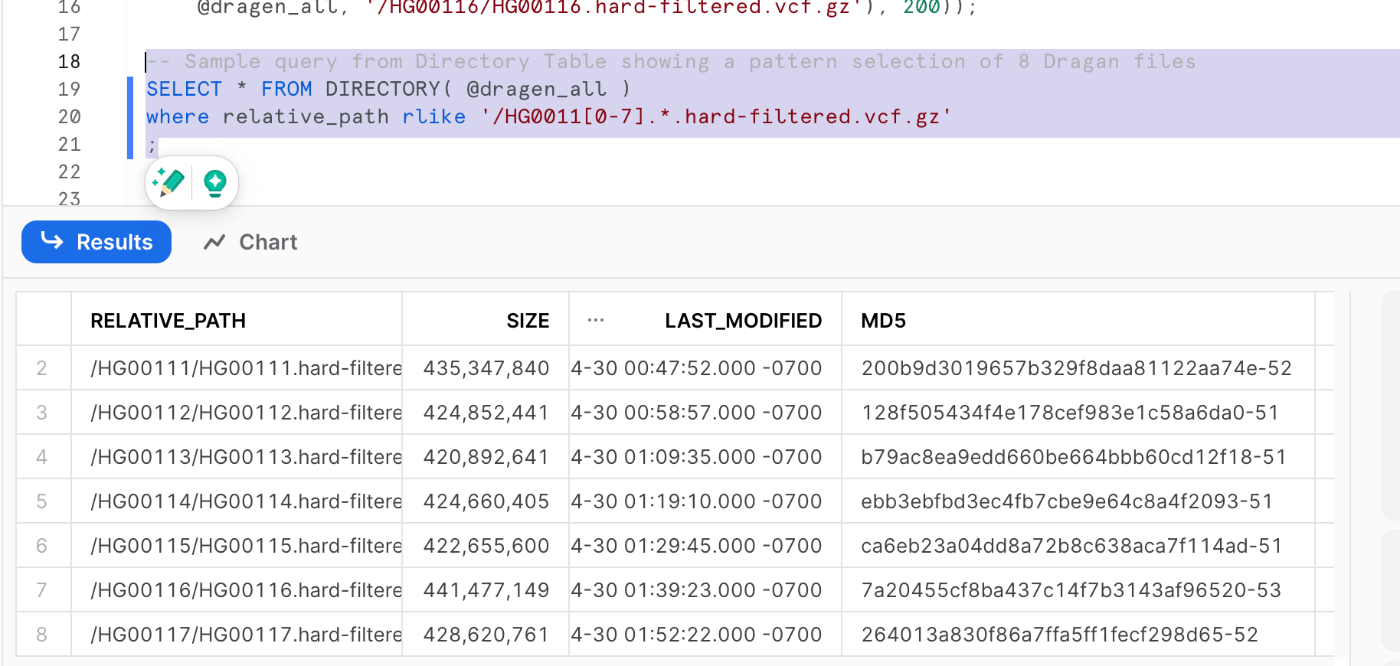
ステージの定義が終わり、解析関数を定義が終わるとクエリを叩けるようになります。"ハードフィルターされた "バリアントとして表現された8つのゲノムのランダムなセットを扱うには、次のようなクエリーでファイルリストを得ることができます。
-- Sample query from Directory Table showing a pattern selection of 8 Dragan files
SELECT * FROM DIRECTORY( @dragen_all )
where relative_path rlike '/HG0011[0-7].*.hard-filtered.vcf.gz'
;

VCFファイルの内容をざっと見るには、次のようなクエリーを使用して最初の200行を取得することができる。
-- Sample query from Directory Table showing a pattern selection of 8 Dragan files
SELECT * FROM DIRECTORY( @dragen_all )
where relative_path rlike '/HG0011[0-7].*.hard-filtered.vcf.gz'
;
特定のSampleIDを含む、Chrom、Pos、Refなどの明確なカラムを含む構造化された結果が表示されます。半構造化フィールドのINFOとSAMPLEVALSには、その場所でのゲノムコールの様々な特徴が表示されます。これらの半構造化フィールドから必要な値を簡単に選択し、操作することができます。
ゲノムバリアントデータを格納するテーブルを作成し、サンプルごとに1バリアントあたり1行でバリアントを表現します。
create or replace table GENOTYPES_BY_SAMPLE (
CHROM varchar,
POS integer,
ID varchar,
REF varchar,
ALT array ,
QUAL integer,
FILTER varchar,
INFO variant,
SAMPLE_ID varchar,
VALS variant,
ALLELE1 varchar,
ALLELE2 varchar,
FILENAME varchar
);
そして最後に、先に定義したVCF解析関数を使用するクエリーをテーブルに投入し、ディレクトリリストへのJOINを介して、目的の各ファイルに適用していきます。(私の環境ではxsサイズで6分15秒でした。)
insert into GENOTYPES_BY_SAMPLE (
CHROM ,
POS ,
ID ,
REF ,
ALT ,
QUAL ,
FILTER ,
INFO ,
SAMPLE_ID ,
VALS ,
ALLELE1 ,
ALLELE2 ,
FILENAME )
with file_list as (
SELECT file_url, relative_path FROM DIRECTORY( @dragen_all )
--where relative_path rlike '.*.hard-filtered.vcf.gz' //select all 3202 genomes
where relative_path rlike '/HG0011[0-7].*.hard-filtered.vcf.gz' //selection of 8 genomes
--where relative_path rlike '/HG00[0-2][0-9][0-9].*.hard-filtered.vcf.gz' //selection of 122 genomes
order by random() //temporary hint to maximize parallelism
)
select
replace(CHROM, 'chr','') ,
POS ,
ID ,
REF ,
split(ALT,','),
QUAL ,
FILTER ,
INFO ,
SAMPLEID ,
SAMPLEVALS ,
allele1(ref, split(ALT,','), SAMPLEVALS:GT::varchar) as ALLELE1,
allele2(ref, split(ALT,','), SAMPLEVALS:GT::varchar) as ALLELE2,
split_part(relative_path, '/',3)
from file_list,
table(ingest_vcf(BUILD_SCOPED_FILE_URL(@dragen_all, relative_path), 0)) vcf
;
データ・プロパティのクエリー例
データから初期特性を得てみましょう。
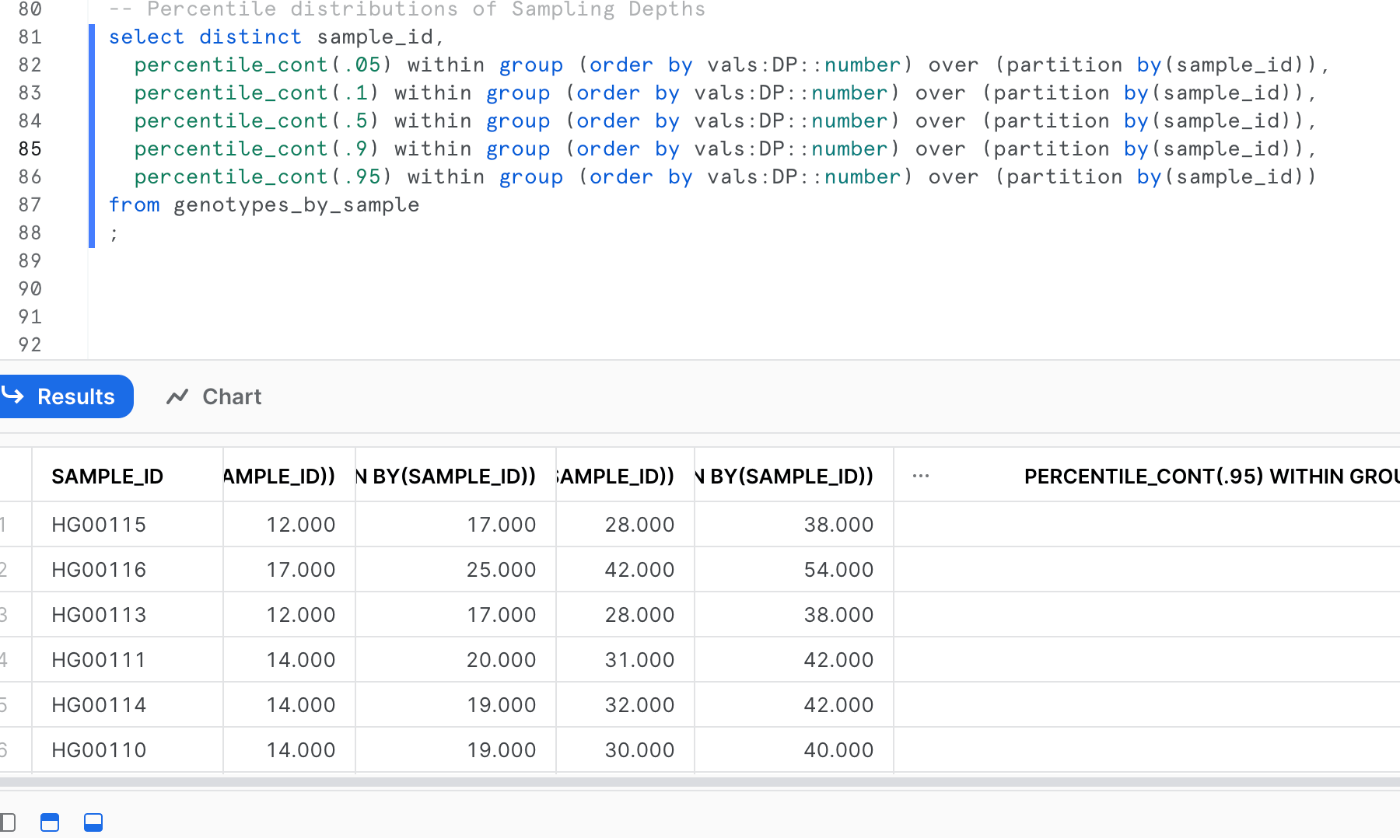
以下のクエリは、サンプルごとのバリアントコールのサンプリング深度(DP)値の分布を要約し、この重要な品質指標の5、10、50、90、95パーセンタイル値を示しています
-- Percentile distributions of Sampling Depths
select distinct sample_id,
percentile_cont(.05) within group (order by vals:DP::number) over (partition by(sample_id)),
percentile_cont(.1) within group (order by vals:DP::number) over (partition by(sample_id)),
percentile_cont(.5) within group (order by vals:DP::number) over (partition by(sample_id)),
percentile_cont(.9) within group (order by vals:DP::number) over (partition by(sample_id)),
percentile_cont(.95) within group (order by vals:DP::number) over (partition by(sample_id))
from genotypes_by_sample
;
VCF(Variant Call Format)データを解析する際の注意点ですが、同じ遺伝子型でも集団間で正規化されていないデータを扱うときには、具体的な対立遺伝子(アレル)の値を直接使用することが推奨されています。
別の例では、各染色体で観察されたバリアントコールの量を、ホモ接合バリアントかヘテロ接合バリアントかで特徴付け、ALLELE1とALLELE2の値を使って決定しています。インジェストしたファイルには、サンプルごとに呼び出されたバリアント(非参照)のみが含まれているので、ホモ接合の参照カウントはすべて0になります。
-- Total Count of variant calls by type, per chrom
select chrom,
sum(case when allele1 = ref and allele2 = ref then 1 else 0 end) as homozref,
sum(case when allele1 != allele2 then 1 else 0 end) as heterozalt,
sum(case when allele1 != ref and allele2 = allele1 then 1 else 0 end) as homozalt
from Genotypes_by_sample
where try_to_number(chrom) is not null
group by 1 order by try_to_number(chrom);

アノテーションと表現型データソースの統合
いよいよ遺伝子型データをアノテーションや表現型情報と結びつけていきます。
ClinVarデータの現在のコピーはAWSによってParquetフォーマットで管理されているので取り込んでいきます。ちなみにClinVarは、アメリカ国立衛生研究所(National Institutes of Health: NIH)が運営する無料の公開データベースで、人間の遺伝変異とその健康への影響(特に遺伝性疾患)に関する情報を集約しています。
まずs3ロケーションを指すステージを定義し、Parquetを構造化テーブルにインジェストします:
create or replace stage clinvar
url = 's3://aws-roda-hcls-datalake/clinvar_summary_variants'
file_format = (type = PARQUET)
;
create or replace table CLINVAR (
chrom varchar(2),
pos number ,
ref varchar ,
alt varchar ,
ALLELEID number ,
chromosomeaccession varchar ,
CLNSIG varchar ,
clinsigsimple number ,
cytogenetic varchar ,
geneid number ,
genesymbol varchar ,
guidelines varchar ,
hgnc_id varchar ,
lastevaluated varchar ,
name varchar ,
numbersubmitters number ,
origin varchar ,
originsimple varchar ,
otherids varchar ,
CLNDISB array ,
CLNDN array ,
rcvaccession array ,
CLNREVSTAT varchar ,
RS number ,
startpos number ,
stoppos number ,
submittercategories number ,
testedingtr varchar ,
type varchar ,
variationid number ,
full_annotation variant
);
insert into CLINVAR
select
$1:chromosome ::varchar(2) chrom,
$1:positionvcf ::number pos,
$1:referenceallelevcf ::varchar ref,
$1:alternateallelevcf ::varchar alt,
$1:alleleid ::number ALLELEID,
$1:chromosomeaccession ::varchar chromosomeaccession,
$1:clinicalsignificance ::varchar CLNSIG,
$1:clinsigsimple ::number clinsigsimple,
$1:cytogenetic ::varchar cytogenetic,
$1:geneid ::number geneid,
$1:genesymbol ::varchar genesymbol,
$1:guidelines ::varchar guidelines,
$1:hgnc_id ::varchar hgnc_id,
$1:lastevaluated ::varchar lastevaluated,
$1:name ::varchar name,
$1:numbersubmitters ::number numbersubmitters,
$1:origin ::varchar origin,
$1:originsimple ::varchar originsimple,
$1:otherids ::varchar otherids,
split($1:phenotypeids::varchar, '|') CLNDISB,
split($1:phenotypelist::varchar, '|') CLNDN,
split($1:rcvaccession::varchar, '|') rcvaccession,
$1:reviewstatus ::varchar CLNREVSTAT,
$1:rsid_dbsnp ::number RS,
$1:start ::number startpos,
$1:stop ::number stoppos,
$1:submittercategories ::number submittercategories,
$1:testedingtr ::varchar testedingtr,
$1:type ::varchar type,
$1:variationid ::number variationid,
$1 ::variant full_annotation
FROM '@clinvar/variant_summary/'
where $1:assembly = 'GRCh38'
order by chrom, pos
;
新たに取り込む(インジェストする)データをGRCh38レファレンスゲノムと一致させ、すでに取り込まれた1000 Genomesデータセットとの整合性を取っていきます。
表現型データについては、各サンプルの地理的起源と性別、およびサンプル間の家族連鎖を記述した「panel」テーブルのみ扱うことが可能です。それでも、ゲノムの結合とサンプルの特性を考察するのに十分な材料です。
create or replace stage population
url = 's3://1000genomes/1000G_2504_high_coverage/additional_698_related/'
file_format = (type = CSV compression = AUTO field_delimiter=' ' skip_header=1)
;
create or replace table panel (
Sample_ID varchar,
Family_ID varchar,
Father_ID varchar,
Mother_ID varchar,
Gender varchar,
Population varchar,
Superpopulation varchar
);
insert into panel
select $2, $1, $3, $4, case when $5 = 1 then 'male' else 'female' end, $6, $7
from '@population/20130606_g1k_3202_samples_ped_population.txt'
;
すべてをまとめる
panelデータとゲノムロケーションデータを統合して興味あるバリアント解析ができるようになりました。それでは具体的な解析を行っていきましょう!
特定の人口統計条件(今回は、イギリスの女性)の人々について、指定されたクロマソーム(クロマソーム10)の特定の位置範囲(100,000から500,000の間)の遺伝子型情報を取得してみます。
-- Example Query: Britain Female Samples for subset of chrom 10
select g.sample_id, chrom, pos, ref, alt, vals:GT::varchar gt,
allele1,
allele2
from genotypes_by_sample g
join panel p on p.sample_id = g.sample_id
where chrom = '10' and pos between 100000 and 500000
and population = 'GBR'
and gender = 'female'
limit 100;
段々と解析データっぽくなってきましたね。
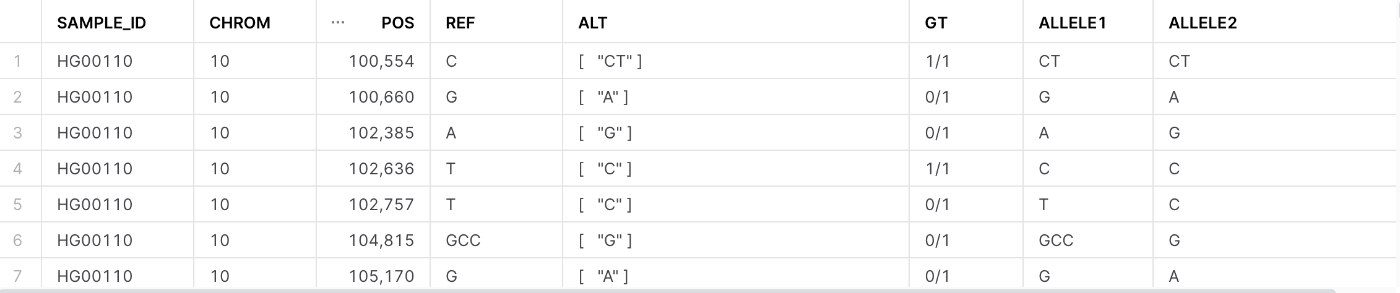
各カラムの解説は以下の通りです。
sample_id - サンプルID。ここで言うサンプルとは、具体的には個々のゲノムサンプルを指します。
chrom - クロマソーム番号。このクエリでは、クロマソーム10のデータを取得します。
pos - クロマソーム上の位置。このクエリでは、クロマソーム10の100,000から500,000の間の位置のデータを取得します。
ref - リファレンス(参照)アリル。通常はヒトゲノムの「標準的な」バージョンに見られる塩基(DNAの文字)です。
alt - 代替アリル。変異が見られる場合の塩基(DNAの文字)です。
gt - 遺伝子型。これは個体がその位置で持っている具体的なアリルのペアを示します。
allele1 と allele2 - 遺伝子型が示す、選択された2つのアリル。
次に遺伝性大腸がんに関連するすべての位置を調べます。染色体や位置を明示的に指定する必要はなく、ClinVarへの結合によって暗黙的にフィルタリングが適用されることに注意してください:
-- Example Query -- locations associated with hereditary colon cancer
select g.chrom, g.pos, g.ref, allele1, allele2, count (sample_id)
from genotypes_by_sample g
join clinvar c
on c.chrom = g.chrom and c.pos = g.pos and c.ref = g.ref
where
array_contains('Hereditary nonpolyposis colorectal neoplasms'::variant, CLNDN)
group by 1,2,3,4,5
order by 1,2,5
;
ClinVarのCLNDNカラムを扱う場合、カラムに複数の値が含まれる可能性があるため、フィルター条件でSnowflakeのARRAY_CONTAINS関数を使用して目的の指標を指定します。次に、バリアントコールのALLELE1またはALLEL2がアノテーションと一致するバリアント値のみを明らかにすることで、クエリを絞り込むことができます。
-- Example Query -- specific variants associated with hereditary colon cancer
select g.chrom, g.pos, g.ref, c.alt clinvar_alt, genesymbol, allele1, allele2, count (sample_id)
from genotypes_by_sample g
join clinvar c
on c.chrom = g.chrom and c.pos = g.pos and c.ref = g.ref
and ((Allele1= c.alt) or (Allele2= c.alt))
where
array_contains('Hereditary nonpolyposis colorectal neoplasms'::variant, CLNDN)
group by 1,2,3,4,5,6,7
order by 1,2,5
;
ゲノムデータの価値を引き出すその他の方法
業界標準のVCFフォーマットで作成されたゲノムデータを、患者データやアノテーションデータと組み合わせて取り込み、結合、解析できるだけでもsnowflakeの柔軟性を感じますが、他にも様々なユースケースに適用できることが考えられます。以下のようなことに応用できると述べられていました。
・選択したデータサブセットをコピーすることなく、共同作業者と安全にロールガバナンス付きで共有する。
・集団全体のデータコレクションを蓄積するために、シーケンスごとに新しいデータをインクリメンタルに取り込む。
・生のゲノムデータセットを「正規化」コレクションに変換する
・GVCFデータソースの取り扱いと高品質レンジリードからのリファレンスコールの導出
・サンプルIDまたはゲノム内の位置による高速検索を可能にするインデックス/クラスタリング技術
・バリアントと表現型を組み合わせたデータに基づいて、機械学習モデルのトレーニングまたはスコアリングの入力としてデータフレームを生成する。
個人的にバイオインフォマティクスする上でsnowflakeに足りないところ
個人的にsnowflakeでバイオインフォマティクスをするには1.パイプライン処理の実行と2.バイオインフォマティクスライブラリのサポートが必要だと思います。
1.パイプライン処理の実行
生命科学の領域では、様々な分析ツールが存在します。それらは単独で使用することも可能ですが、より複雑な分析や解析を行うためには、これらのツールを組み合わせて一連のパイプラインを構築し、その上で処理を行うことが一般的です。RNA-seqデータの解析を例にとってもfasterq-dump(sra-toolkit)→QC(FastQC)→リードアラインメント(STAR、Hisat2)→リードカウント(salmon)→正規化(DESeq2やedgeR)→下流解析(ggplot2やenrichment解析)などあります。snowflake上にサーバーレスに実行できるコンピューティングリソースをキャパシティ課金で使えるようになると柔軟な解析ができると思います。加えてNextflowやKNIMEなどのパイプライン処理言語がサポートされるのも重要です。
今年のsnowflakesummitで発表されたsnowpark container servicesだとk8s上でコンテナが動かせるのでDockerを使ってLinux環境を構築すれば、バイオインフォマティクスの分析ツールを自由自在に扱うことが可能になります。これにより、自身のニーズに最適な環境を構築し、最新の科学的手法を活用して研究を進めることが可能になるかもしれません。
2.バイオインフォマティクスライブラリのサポート
snowflakeでpythonを使えるsnowpark for pythonですが、バイオインフォマティクス系のライブラリはあまり充実しておりません。Snowflake conda channelではbio系のライブラリといったらbiopythonぐらいでした。
残念ながら、single cell RNA-seqライブラリであるscanpyはサポートされていないみたいです。シングルセルデータは超巨大データであるためsnowflake上でさばけると処理が効率化されます。snowsight上に10X genomics フォーマットサンプルのUMAPプロットを表示させる未来を個人的には心待ちにしています。知らない方のために、私が過去に解析したpbmcデータに免疫細胞のアノテーションを自動付与したUMAPプロットを例として載せておきます。

バイオ系でもpythonは人気ですが、Rも一定数の人気があります。相互でできることが異なっており、依然としてRでしかできない解析も多く存在することから、バイオインフォマティシャンの中にはPython/Rの二刀流を取っている方も存在します。先程紹介したsnowpark container servicesではRのロゴがあったのでRが使えるようになることも期待しています。
しかしできることも多い
しかし、snowflake上でできるインフォマティクスも現状の段階で十分にあります。ハンズオンで取り上げた公共のS3を外部ステージとして統合して取り込むのは、複雑なETL処理やAPIでTSVファイルを引っ張ってくるといった煩雑さから開放してくれます。またバイオデータは臨床検体といった患者のプライバシーに当たるデータを取り扱う機会も多く、このあたりはsnowflakeのデータマスキングやセキュアなデータ共有機能が活かせるポイントです。
またバイオデータはその複雑性から往々にして半構造化データとなっている場合も多く、snowflakeだと半構造化データのまま取り込んでクエリできるのは魅力的な要素と言えます。このあたり、SAM/BAMといった次世代シーケンサーのファイルに対して高速で解析をかけるといったユースケースも近い将来では可能になりそうです。
また最近のsnowflakeはstreamlit周りが充実してきました。一度ゲノムデータをsnowflake上のテーブルに構築すれば、streamlitによる可視化でグラフデータの取得が可能です。例えば以下はcovid19の重症度に応じた各遺伝子発現量(リードカウント)を箱ひげプロットにて可視化しています。RNA-seqデータのような巨大データを格納→streamlitで可視化といった流れは有効な手段となりえます。
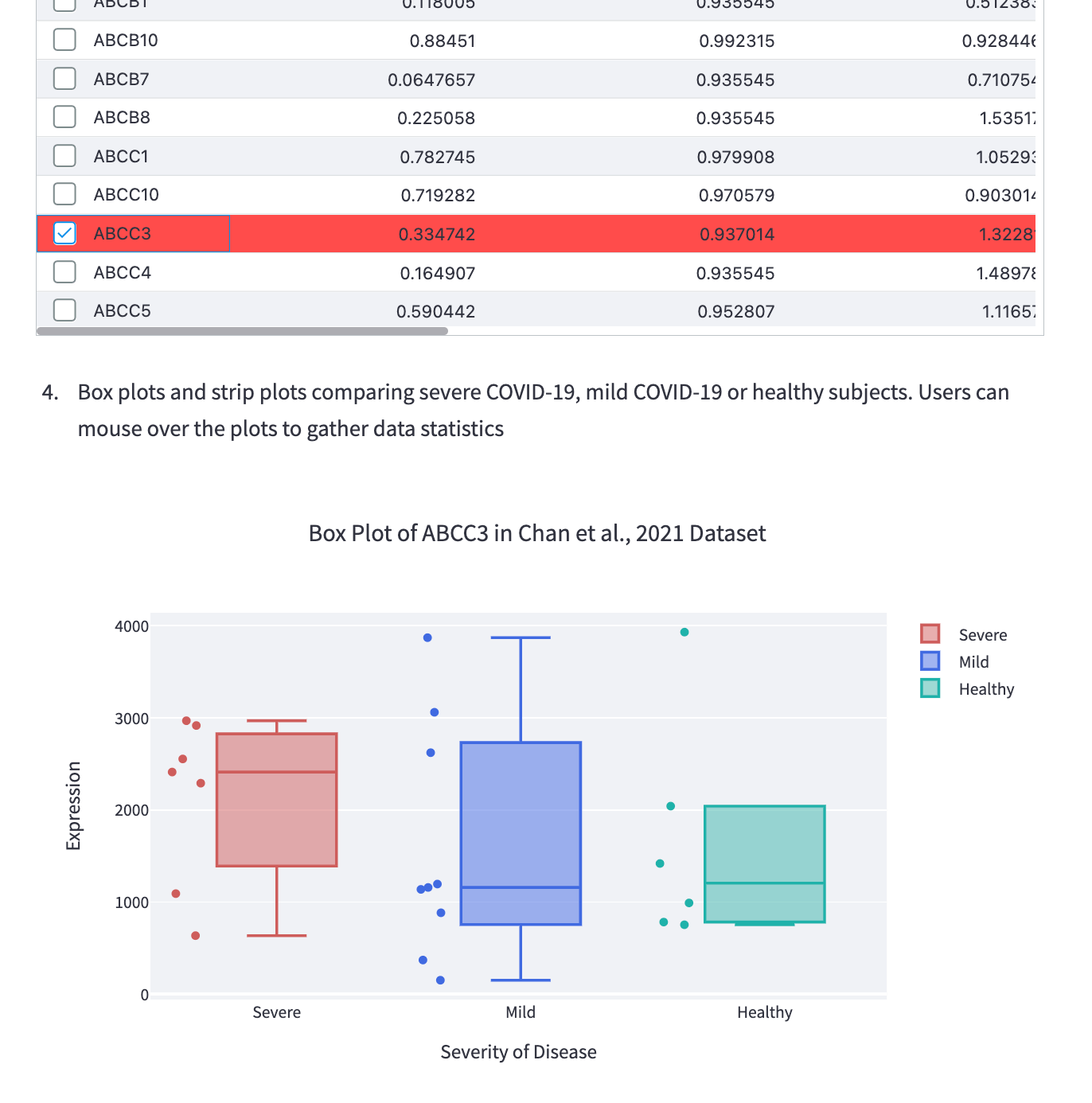
終わりに
今回はsnowflakeで行うバイオインフォマティクスの可能性を探求してみました。ちなみにsnowflakeをdatawarehouseとして使って解析した論文はあるかPubmed検索してみましたがありませんでした。
個人的にはGigaScienceあたりのジャーナルでsnowparkを使ったオミクス解析や高速配列検索システムあたりで論文化するのも面白そうだなと感じてます。
Discussion