AdaptyvBioタンパク質デザインコンペ参戦記
注意書き: 本記事の執筆には誤字脱字などの文章校正以外に 生成AIを利用していません。
概要
本記事はAdaptyvBio主催のProtein Design Competitionの記録です。2025年2月現在までで、ラウンド1と2が開催されており、ラウンド2に参加しました。両ラウンドともに、EGFRに結合しEGFリガンドの結合を阻害するバインダーの設計が課題となっています。またラウンド1では合計700のバインダーデザインが提出され、200のデザインがWET実験に進みました。それらの結果はGitHub上で公開されています。
コンペティションの基本的なルールとしては、天然アミノ酸から構成される配列であること、250残基以内であること、シングルチェーンであること、公開されている配列から最低10アミノ酸以上の編集距離があることです。編集距離については、新規のアミノ酸配列であることを保証するものです。また、本コンペティションはCET中央ヨーロッパ時間で2024年11月4日まで開催されました。
EGFR結合バインダー設計
タンパク質のバインダー設計とはある標的タンパク質の特定部位に高い親和性と選択製で結合する新規のタンパク質(バインダー)を設計することです。以下はBennettらの論文内の図なのですが、バインダー設計のイメージを掴めるかと思います。

Fig. 1: Monomer and protein complex structure prediction metrics distinguish previously designed binders from non-binders.[1]
コンペティションでは、EGFRに結合し、EGFリガンドの結合を阻害するバインダーの設計が求められています。EGFRのの配列と立体構造は以下の通りです。配列については、EGFRのリガンド結合部位に結合するエピトープが重要であるため、この部位に結合するデザインが求められます。EGFリガンドは、ドメインⅠの11-13, 15-18,ドメインⅢの356, 440-441がエピトープとして知られています。また立体構造については、3D立体構造図の緑色で絵示されているのがEGFRで、紫(mutant heavy chain)とオレンジ色(mutant light chain)の複合体がCetuximabです。CetuximabはEGFRのドメイン3に結合することがわかります。
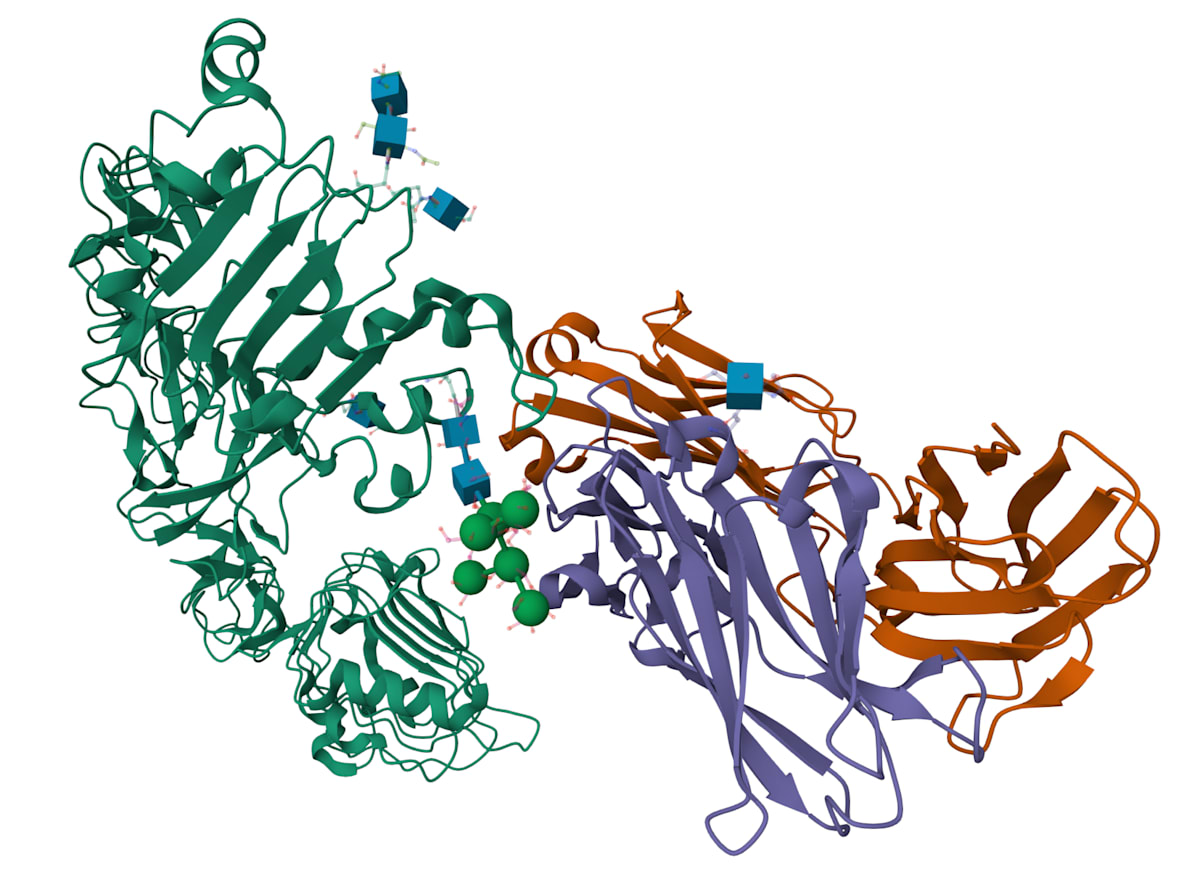
EGFR and Cetuximab 3D structure
以下に、EGFR、EGF、およびCetuximab(既存のEGFR阻害剤)について簡単に説明します。
-
EGFR(上皮成長因子受容体)
EGFRは細胞膜に存在する重要な受容体で、細胞の成長、分化、生存に深く関与しています。通常、EGFなどのリガンドが受容体の外部ドメインに結合すると、EGFRは二量体化して活性化され、細胞内シグナル伝達経路(Ras/MAPK、PI3K/Akt、STATなど)を起動します。がん細胞では、EGFRの過剰発現や特定の変異によって、これらのシグナルが恒常的に活性化され、異常な細胞増殖が促進されるため、腫瘍形成や進行に寄与しています。

Figure 3: The effect of radiation and chemotherapy on EGFR signalling.[2]
-
EGF(上皮成長因子)
EGFはEGFRの天然リガンドであり、受容体の外部リガンド結合領域に結合することで、EGFRの二量体化と活性化を引き起こします。正常な細胞では、この結合により細胞の増殖や分化が適切に制御されますが、がん細胞ではEGFによる過剰な刺激が細胞増殖シグナルを強め、腫瘍の形成や進行に関与することがあります。
-
Cetuximab
Cetuximabは、EGFRを標的としたモノクローナル抗体医薬品です。EGFRの外部リガンド結合部位に結合することで、EGFの結合を阻害し、受容体の二量体化およびその後のシグナル伝達を抑制します。その結果、がん細胞における過剰な増殖シグナルが遮断され、腫瘍の進行が抑えられます。
評価指標
本コンペティションでは、指定された評価指標に基づいてスコアが計算され、リーダーボードに掲載されます。そしてそのうちの上位100のバインダーデザインがWET検証に進みます。評価指標の計算コードはこちらで公開されています。ラウンド2ではPAE, IPTM,ESM2 log-likelihoodの3つの指標が使用されました。
-
PAE (Predicted Aligned Error)
PAEはAlphaFoldが予測したタンパク質構造の信頼性を示す指標です。具体的には、モデル内の任意の2つの原子間の位置予測誤差を表します。PAEが低いほど、予測された構造の信頼度が高いことを意味します。
-
IPTM (Interface Predicted TM-score)
IPTMはバインダーとEGFRの界面の品質を評価する指標です。TM-scoreをベースにしており、二つのタンパク質の界面が正確にモデル化されているかを0から1の値で表します。1に近いほど高品質な界面を示します。長さ
-
ESM2 log-likelihood
ESM2[1:1]はFacebookが開発した大規模言語モデルで、タンパク質配列の自然さを評価します。ESM-2 log-likelihoodはデザインされた配列が自然なタンパク質配列にどれだけ似ているかを示し、高い値ほど配列が生物学的に妥当である可能性が高いことを意味します。
-
Metrics
MetricsはPAE, IPTM, ESM2 log-likelihoodのランクの平均値で、リーダーボードに掲載されるスコアとして使用されます。Metricsが低いほど高いスコアを獲得できます。
-
r^{\uparrow/\downarrow}(\cdot) -
\text{iPTM} -
\text{PAE} -
\text{pLL}
備考セクションにて、精度評価リポジトリより実際にリーダーボードの計算に使用される実装を示します。バインダーデザインの提出前にこちらの実装を使用して事前にスコアを計算し、ある程度のリーダーボード上の位置を予測することができます。ColabFold[5]で事前にバインダーデザインから立体構造予測を行い、その結果をダウンロードしておく必要があります。リーダーボードのスコア計算にもColabFoldで精度評価(各サンプル3回ずつ)が行われており、ローカル環境での評価と概ね一致していました。
 Colabfoldの設定画面 Colabfoldの設定画面
|
 ColabFoldで予測した複合体の立体構造 ColabFoldで予測した複合体の立体構造
|
 予測した複合体のチェーンとplDDTのカラーリング 予測した複合体のチェーンとplDDTのカラーリング
|
 配列のカバレッジプロット 配列のカバレッジプロット
|
BindCraft
今回はBindCraftというバインダーデザインのパイプラインを使用しました。というのも、ラウンド1では、Martin氏がBindCraftでデザインしたバインダーが1st placeを獲得し、その後のWET実験でも最も強い結合親和性を示しました (Martin氏はBindCraftの著者です)。RFDiffusion[6]やRossetta[7]などの手法も有用だと思いましたが、今回の評価指標の内、pae interactionとipTMは、AlphaFold2[8]を使用して計算されるので、より多くのデザインをWET検証に進めるためには、AlphaFold2のBackboneを使用するBindCraftが適していると考えました。ESM2 loglikelihoodは、配列全体の総和で計算されるので、配列長が大きいほど相対的にリーダーボード上のスコアは低くなります。リーダーボード上でより上位に行くためには既知のバインダーを最適化する手法の方が有利ですが、3つの評価指標を最適化することとWET実験で高い結合親和性を目指すことは必ずしもイコールではないので、WET実験での期待値が高い手法を採用しました。

BindCraft binder design pipeline[9]

Comepetition overview and winners
簡単にBindCraftの実行方法を簡単に紹介します。User friendly and accurate binder design pipelineと述べられている通り、以下のように容易にパイプラインを実行できます。またGoogle Colaboratory上のノートブックも公開されています。ただし注意点として、32GB以上のGPUメモリが推奨されています。特に入力配列が長い場合は、実行可能な長さにトリミングすることも推奨されています。入力配列が長いと、単純にGPUメモリの使用量が増加するだけでなく、探索空間が広すぎてフィルターを通過するのが難しくなり、実行時間が長くなります。今回のコンペでも、EGFR配列全体(621残基)を入力するとH100(80GB)で24時間実行しても、デフォルトのフィルターを通過したデザインはありませんでした。今回はドメイン1とドメインⅢのそれぞれにトリミングしてパイプラインを実行しました。
# clone BindCraft repository
git clone https://github.com/martinpacesa/BindCraft.git
cd BindCraft
# Install BindCraft
bash install_bindcraft.sh --cuda '12.2' --pkg_manager 'mamba' # or conda
mamba activate BindCraft
# run pipeline
python -u ./bindcraft.py
--settings './settings_target/egfr.json'
--filters './settings_filters/default_filters.json'
--advanced './settings_advanced/default_4stage_multimer.json'
{
"design_path": "./designs/EGFR/",
"binder_name": "EGFR",
"starting_pdb": "./data/EGFR.pdb", # target protein structure
"chains": "A",
"target_hotspot_residues": "A356,A440-441",
"lengths": [100, 120],
"number_of_final_designs": 10
}
BindCraftのデフォルトのフィルター設定では、基本的にαヘリックスのバインダーが生成されます。ここもフィルターの設定でチューニング可能で、実際にMartin氏らはBindCraftで生成したβシートのデザインをラウンド2では提出していました(ラウンド1の時に作成したデザインのようです)。
DRY実験結果
全てのバインダーデザインをNocoDBで公開しています。今回は提出しませんでしたが、リラックスされた配列空間において勾配降下法を用いた反復的な最適化(ハルシネーション)によるde novoデザイン手法であるRSO[10]を使用して生成されたデザインも含めています。"リラックスされた"とは、本来離散的なアミノ酸配列を連続的な表現することで、離散的な探索空間の制約を緩和する手法のようです。実際にはModalで実行できるようにしたCorey氏の実装を使用しています(GitHub)。
ターゲット配列のトリミングする領域と生成する配列長を変えながら生成しており、生成された配列長と評価指標の分布は上右図の通りです。リーダーボード上では、以下の通り、最高で200位、最低で652位という結果となりました。今回のコンペでは、やはりPLL(ESM2 loglikelihood)によるランクの変動が大きかったように思います。既知の配列であるほど当然PLLは高くなるので、上位100にランクインしているデザインの多くは既知のバインダーを最適化したものでした。そう考えるとde novoデザインは少し不利なように聞こえますが、WET実験で3rd placeを獲得した手法は3つの評価指標を上手く最適化しており、個人の戦略次第なところではあります。

WET実験結果
当初はDRY実験のリーダーボード上位100のバインダーデザインと、AdaptyvBioによって選ばれた100のバインダーデザインがWET実験に進む予定でした。しかしラウンド2は想定よりも多くの参加者がいたため、AdaptyvBioによって選ばれるデザイン数が300に増え、合計で 400 のバインダーデザインがWET検証されました。AdaptyvBioによって選ばれる基準は、デザイン手法の新規制や独創性など、リーダーボードのスコア以外の要素が考慮されるようで、各チーム1から5のデザインが選ばれました。
今回WET実験に進んだデザインは次の3デザインです。
そのうちEGFR_l110_s478327_mpnn7_model2はCetuximabには及ばなかったものの、EGFリガンドよりも強く結合することが確認されました。

提出したデザインの内、EGFR結合親和性を示したバインダーデザインとその結果

WET検証後のリーダーボード (8位から14位)
提出したデザインの詳細な実験結果はコンペティションホームとGitHubで公開されています。WET検証された400デザイン中14番目に強い結合親和性を示しており、これは110名の全参加者のうち8番目の結果でした。上位3チームにはNeurlPS 2024のAIDrugX Workshopでのプレゼンの機会が与えられ、さらに上位5チームには追加で10個のアッセイを送ることができます。また上位10チームにはAdaptyvBioのグッズが送られるようです。また、ラウンド2の詳細はこちらにも公開されています。
参考資料
- AdaptyvBio The Protein Design Competition
- AdaptyvBio The Protein Design Competition (Round2 Results)
- Predicting the Effects of Mutations on Protein Function with ESM-2
- Section 3: Interpreting results from AlphaFold Server
- Integration of EGFR inhibitors with radiochemotherapy
- BindCraft: one-shot design of functional protein binders
備考
- BindCraft (Martin氏のTweerより)
- EGFRのアミノ酸配列 (621残基)
LEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVVLGNLEITYVQRNYDLSFLKTIQEVAGYVL
IALNTVERIPLENLQIIRGNMYYENSYALAVLSNYDANKTGLKELPMRNLQEILHGAVRFSNNPAL
CNVESIQWRDIVSSDFLSNMSMDFQNHLGSCQKCDPSCPNGSCWGAGEENCQKLTKIICAQQCSGR
CRGKSPSDCCHNQCAAGCTGPRESDCLVCRKFRDEATCKDTCPPLMLYNPTTYQMDVNPEGKYSFG
ATCVKKCPRNYVVTDHGSCVRACGADSYEMEEDGVRKCKKCEGPCRKVCNGIGIGEFKDSLSINAT
NIKHFKNCTSISGDLHILPVAFRGDSFTHTPPLDPQELDILKTVKEITGFLLIQAWPENRTDLHAF
ENLEIIRGRTKQHGQFSLAVVSLNITSLGLRSLKEISDGDVIISGNKNLCYANTINWKKLFGTSGQ
KTKIISNRGENSCKATGQVCHALCSPEGCWGPEPRDCVSCRNVSRGRECVDKCNLLEGEPREFVEN
SECIQCHPECLPQAMNITCTGRGPDNCIQCAHYIDGPHCVKTCPAGVMGENNTLVWKYADAGHVCH
LCHPNCTYGCTGPGLEGCPTNGPKIPS
- ColabFoldの出力ディレクトリからiPAE, ipTM, PLLを計算するPythonスクリプト
import os
import json
import math
import numpy as np
import torch
import esm
import click
def get_metrics(
output_folder, sequence_name, target_length=621
): # the EGFR sequence has 621 residues
files = os.listdir(output_folder)
pae_file = f"{sequence_name}_predicted_aligned_error_v1.json"
with open(os.path.join(output_folder, pae_file), "r") as f:
pae = np.array(json.load(f)["predicted_aligned_error"])
binder_len = (
len(pae) - target_length
) # note that we are assuming that the binder always comes first in the sequence!
pae_interaction = (
pae[:binder_len, binder_len:].mean() + pae[binder_len:, :binder_len].mean()
) / 2
scores = None
for file in files:
if file.startswith(f"{sequence_name}_scores_rank_001"):
with open(os.path.join(output_folder, file), "r") as f:
scores = json.load(f)
break
if scores is None:
raise ValueError("No scores file found")
iptm = scores["iptm"]
return iptm, pae_interaction
def compute_pll(sequence):
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
model, alphabet = esm.pretrained.esm2_t33_650M_UR50D()
batch_converter = alphabet.get_batch_converter()
model.eval()
model = model.to(device)
data = [("protein", sequence)]
batch_converter = alphabet.get_batch_converter()
*_, batch_tokens = batch_converter(data)
log_probs = []
for i in range(len(sequence)):
batch_tokens_masked = batch_tokens.clone()
batch_tokens_masked[0, i + 1] = alphabet.mask_idx
with torch.no_grad():
token_probs = torch.log_softmax(
model(batch_tokens_masked.to(device))["logits"], dim=-1
)
log_probs.append(token_probs[0, i + 1, alphabet.get_idx(sequence[i])].item())
return math.fsum(log_probs)
@click.command()
@click.option(
"--target_length", default=621, help="Length of the target protein (in amino acids)"
)
@click.argument("output_folder")
@click.argument("sequence_name")
@click.argument("aa_sequence")
def main(output_folder, sequence_name, target_length, aa_sequence):
iptm, pae_interaction = get_metrics(output_folder, sequence_name, target_length)
pll = compute_pll(aa_sequence)
print(f"IPTM: {iptm}")
print(f"PAE interaction: {pae_interaction}")
print(f"PLL: {pll}")
if __name__ == "__main__":
main()
- Top10の参加者に送られるAdaptyvBioのグッズ

Adaptyv Bio & Polaris Merch Pack
-
Alexander Rives, Joshua Meier, Tom Sercu Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences (2021) https://doi.org/10.1073/pnas.2016239118 ↩︎ ↩︎
-
Nyati, M., Morgan, M., Feng, F. et al. Integration of EGFR inhibitors with radiochemotherapy. Nat Rev Cancer 6, 876–885 (2006). https://doi.org/10.1038/nrc1953 ↩︎
-
Richard Evans, Michael O’Neill, Alexander Pritzel, Natasha Antropova, Andrew Senior, Tim Green, Augustin Žídek, Russ Bates, Sam Blackwell, Jason Yim, Olaf Ronneberger, Sebastian Bodenstein, Michal Zielinski, Alex Bridgland, Anna Potapenko, Andrew Cowie, Kathryn Tunyasuvunakool, Rishub Jain, Ellen Clancy, Pushmeet Kohli, John Jumper, Demis Hassabis Protein complex prediction with AlphaFold-Multimer
bioRxiv 2021.10.04.463034; doi: https://doi.org/10.1101/2021.10.04.463034 ↩︎ -
Bennett, N.R., Coventry, B., Goreshnik, I. et al. Improving de novo protein binder design with deep learning. Nat Commun 14, 2625 (2023). https://doi.org/10.1038/s41467-023-38328-5 ↩︎
-
Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682 (2022). https://doi.org/10.1038/s41592-022-01488-1 ↩︎
-
Watson, J.L., Juergens, D., Bennett, N.R. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023). https://doi.org/10.1038/s41586-023-06415-8 ↩︎
-
Minkyung Baek et al. ,Accurate prediction of protein structures and interactions using a three-track neural network.Science373,871-876(2021).DOI:10.1126/science.abj8754 ↩︎
-
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2 ↩︎
-
Martin Pacesa, Lennart Nickel, Joseph Schmidt, Ekaterina Pyatova, Christian Schellhaas, Lucas Kissling, Ana Alcaraz-Serna, Yehlin Cho, Kourosh H. Ghamary, Laura Vinué, Brahm J. Yachnin, Andrew M. Wollacott, Stephen Buckley, Sandrine Georgeon, Casper A. Goverde, Georgios N. Hatzopoulos, Pierre Gönczy, Yannick D. Muller, Gerald Schwank, Sergey Ovchinnikov, Bruno E. Correia BindCraft: one-shot design of functional protein binders bioRxiv 2024.09.30.615802; doi: https://doi.org/10.1101/2024.09.30.615802 ↩︎
-
Christopher Frank et al. ,Scalable protein design using optimization in a relaxed sequence space.Science386,439-445(2024).DOI:10.1126/science.adq1741 ↩︎
Discussion