Amberの金属表面MDのチュートリアルをGROMACSで
Amberのチュートリアル一覧を眺めていると、金属表面のペプチドというものがありました。

(出典:https://ambermd.org/tutorials/advanced/tutorial27/pro_metal.php )
Amberでできるということは、ParmEDで変換すればGROMACSでできるということです(過言)。ということでやってみます。
対象のチュートリアル
Amber Totorials にある Material systems modeling tutorial に、A Protein System at FCC Ag Metal Surface という、金属表面にペプチドを配置しMDを実行するチュートリアルがあります。
この記事はこれを日本語化しつつ、自分のやりやすいように変え、最終的にGROMACSでMDを流すところまで改造していこうと思います。
金属表面の古典MDが手札にあればだいぶいろいろできそうです。ということでこの機会にチャレンジしてみます。
準備
アプリケーションとかライブラリとか
Amberチュートリアルですので、AmberToolsが必要です。GROMACSへの変換にParmedを使いましたので、Pythonありでビルドしましょう。
チュートリアルではVMDを使って分子を操作しているのですが、今回この記事ではASEを使っています。 pip, conda, または apt でインストールしましょう。
あとは適当なバージョンの GROMACS。この記事では 2025.1をビルドして使いました。
モデルと力場ファイル
金属表面ということで、結晶状態の金属原子に対する力場が必要になります。これはAmberToolsとかGROMACSには同梱されてないのですが、INTERFACE-MDという表面力場を作るプロジェクトチームあって、そこがパラメータを公開しています。
このページに行き、中ほどの「Download latest release (1.5)」というリンクを踏んでZIPファイルをダウンロードします。この中にはLammpsによるパラメータとそれに対応する金属の結晶構造ファイルが同梱されています。構造ファイルはcifみたいなやつですが、これはASEで読み込めます。
問題はパラメータで、これは普通には変換できません。ということで、Amberのチュートリアルのページをよく見ると、
このページの一番下の段落が、INTERFACEの力場をAmber向けに変換したこと、これはあくまでテスト用でちゃんとしたプロジェクトにはよく確認してから使ってほしいという注釈があります。この文章中に「interface_v1_5.dat, fcc_metals.lib, and leaprc.interface_v1_5」というところがあり、これがAmber用パラメータファイルのダウンロードリンクになります。これを用意しておきます。
手順
構造pdbファイル作成
金属クラスタの表面にペプチドが乗っている状態のpdbファイルを作ります。
適当な作業ディレクトリを準備し、そのディレクトリに準備の節で説明したINTERFACEのファイル
interface_ff_1_5.zipinterface_v1_5.datfcc_metals.libleaprc.interface_v1_5
がある状態とします。まずzipファイルの解凍から。
unzip interface_ff_1_5.zip
INTERFACE_FF_1_5 というフォルダができます。
続いてペプチドの構造を作ります。以下の内容を seq.in に保存します。または thttps://ambermd.org/tutorials/advanced/tutorial27/files/seq.in からダウンロードします。
source leaprc.protein.ff14SB
mol = sequence {ACE ALA ALA ALA ALA ALA ALA ALA NME}
savepdb mol ace-ala7-nme.pdb
quit
これを以下のように tleap に使うとペプチドの構造ができ、ace-ala7-nme.pdb ができます。
tleap -f seq.in
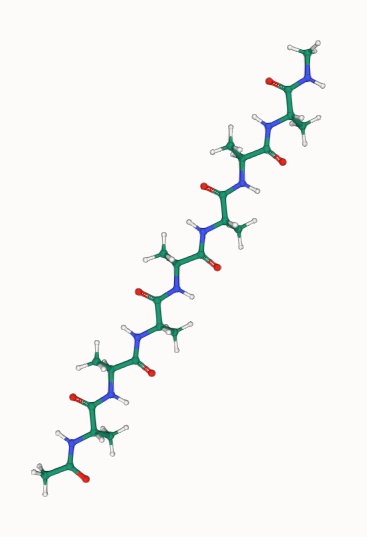
これでペプチドの準備はできました。続いて銀クラスタの準備です。
次の作業はチュートリアルではVMDでやるように書いていますが、ここではASEを使います。 pythonで以下のスクリプトを実行します。pyファイルを作るもよし、pythonの対話環境で1行ずつ入れていくもよし。
from ase.io import read, write
atoms = read("INTERFACE_FF_1_5/MODEL_DATABASE/METALS/ag_cell_P1_100.car")
atoms.write("Ag_Cell_P1_100.pdb")

Ag_Cell_P1_100.pdb を以下のように編集します。原子種とResidue name、Residue indexが変わっています。
CRYST1 4.086 4.086 4.086 90.00 90.00 90.00 P 1
MODEL 1
ATOM 1 Ag0 Ag0 1 0.000 0.000 0.000 1.00 0.00 AG
ATOM 2 Ag0 Ag0 2 0.000 2.043 2.043 1.00 0.00 AG
ATOM 3 Ag0 Ag0 3 2.043 0.000 2.043 1.00 0.00 AG
ATOM 4 Ag0 Ag0 4 2.043 2.043 0.000 1.00 0.00 AG
ENDMDL
AmberToolsを使って、Ag単位セルを8x8x8に並べてクラスタを作ります。4x8x8x8=2048原子の系になります。
PropPDB -p Ag_Cell_P1_100.pdb -o Ag_s100_2048.pdb -ix 8 -iy 8 -iz 8
これで銀クラスタもできました。

これに cat コマンドでペプチドのpdbファイルをくっつけます。そしてまた AmberToolsの pdb4amber で、残基や原子タイプなどを良い感じにします。
cat ace-ala7-nme.pdb Ag_s100_2048.pdb > pro_metal.pdb
pdb4amber -i pro_metal.pdb -o pro_metal_renum.pdb
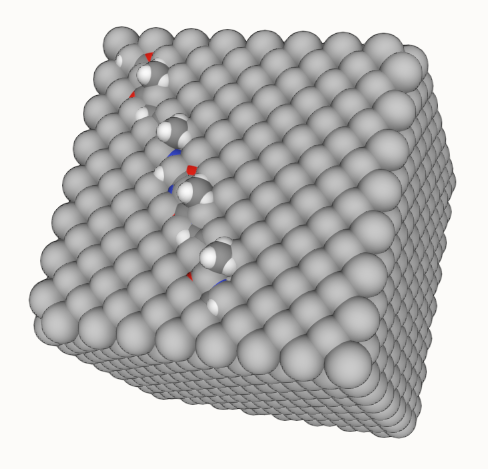
ただこの状態では金属クラスタにペプチドがめり込んでいますので、位置調整をします。チュートリアルではVMDを使って編集していますが、ここでもASEを使います。プログラム的にやった方がどれだけ動かしたかを記録・再現できますし。
以下のpythonコードを実行します。ペプチドを動かす量はチュートリアルで見える途中経過pdbファイルを見てなんとなく決めました。
import numpy as np
from ase.io import read, write
atoms = read("pro_metal_renum.pdb")
mask = np.logical_not(atoms.symbols == "Ag")
pos = atoms.get_positions()
pos[mask, :] += np.array([0, 1.75, -3.166])
atoms.set_positions(pos)
atoms.write("pro_metal_adjust.pdb")
しかし、この pro_metal_adjust.pdb はASEの基準で原子名・残基名が設定され、これがAmber力場を割り当てるのには向いていません。一方で位置調整前の pro_metal_renum.pdb は良い感じですので、これをとってきます。PDBファイルのデータ行でいうと3,4,5番目のカラム、13~26文字目です。
# 適切な atom type や resname, residx を取得
atomtype_resnames = []
with open("pro_metal_renum.pdb", "r") as f:
for L in f.readlines():
if L.startswith("ATOM") or L.startswith("HETATM"):
atomtype_resnames.append(L[12:26])
# 位置修正したpdbファイルのデータ行を読み込み、書き換え
lines = []
with open("pro_metal_adjust.pdb") as f:
iatom = 0
for L in f.readlines():
if L.startswith("ATOM"):
L = L[:12] + atomtype_resnames[iatom] + L[26:]
iatom += 1
lines.append(L)
# 保存
with open("pro_metal_adjust.pdb", "w") as f:
for L in lines:
f.write(L)
これで、 pro_metal_adjust.pdb が以下のような構造になります。これがMDの初期構造となります。

力場トポロジーファイル作成
まずはAmberToolsで作ってしまいます。以下の内容を pro_metal_tleap.in に保存するか、 https://ambermd.org/tutorials/advanced/tutorial27/files/pro_metal_tleap.in からダウンロードします。
source leaprc.protein.ff14SB
source leaprc.interface_v1_5
com = loadpdb pro_metal_adjust.pdb
saveamberparm com pro_metal.prmtop pro_metal.inpcrd
quit
tleap -f pro_metal_tleap.in
を実行すると、 pro_metal.prmtop と pro_metal.inpcrd ができます。ここまでがAmber チュートリアルの内容で、チュートリアルではここからAmberでのMDになります。
GROMACSへ
これをGROMACS用に変換します。以下のコードをpythonで実行します。
import parmed as pmd
system = pmd.load_file("pro_metal.prmtop", "pro_metal.inpcrd")
system.save("pro_metal.top", format="gromacs")
system.save("pro_metal.gro")
これで pro_metal.top と pro_metal.gro ができます。
ただ、なぜか変換の過程で周期境界の長さが変わってしまっている可能性がありますので、pro_metal.gro の最終行を
3.2688 3.2688 8.000
とします。最初二つの 3.2688 は pro_metal_adjust.pdb から、最後の 8.000 はz軸方向を一気に伸ばして真空の領域を作り、銀結晶表面環境としたいためです。ほんとは gmx editconf とかで書き換えるべきかもしれない。
ここまでいろいろ作業ファイルができてディレクトリが散らかってきたので、ディレクトリを分けます。
mkdir gromacs_md
mv pro_metal.top pro_metal.gro gromacs_md
cd gromacs_md
位置拘束
MD中に銀結晶が崩壊しては困るので、位置に調和振動子ポテンシャルを掛けておきます。これはチュートリアルのamberインプットにおける ntr=1, restraintmask=':10-2057', restraint_wt=20.0, に対応することを行います。
まず、以下の内容を posres_metal.itp に保存します。
[ position_restraints ]
; i funct fcx fcy fcz
1 1 8368 8368 8368
ここで 8368 というのはチュートリアルのインプットファイルでは 20 kcal/mol/A^2 というばね定数の力を掛ける設定でしたので、これをGROMACSの単位系の kJ/mol/nm^2 に変換したものになります。
これのフォーマットに自信がなければ gmx genrestr コマンドの結果を参考にするとよいでしょう。
つづいて、pro_metal.top の [ atoms ] で Ag0 を定義しているセクションと、[ system ] のセクションの間に以下を追加します。要は Ag0 に関する拘束ですので、それの [ moleculetype ] 関係のセクション最後に追加しているイメージです。
# ifdef POSRES
# include "posres_metal.itp"
# endif
中身が3行程度だから include とかせず直接書いてもよかったかもしれない。
MD実行
これで準備が整いました。まずはエネルギー最小化で緩和します。以下の内容を em.mdp に保存します。
define = -DPOSRES
; minim.mdp - used as input into grompp to generate em.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
cutoff-scheme = Verlet ; Buffered neighbor searching
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
どこかのサンプルをそのままとってきたのでコメントそのままです。これでコンパイルと実行します。
gmx grompp -f em.mdp -p pro_metal.top -c pro_metal.gro -o em.tpr -r pro_metal.gro
gmx mdrun -deffnm em
grompp の最後の -r pro_metal.gro は位置拘束のための参照位置の指定ファイルです。たぶんこのファイルで指定している位置を中心に、 posres_metal.itp で指定したばね定数のポテンシャルがかかっています。
続いて、そしてこの記事では最後に、NVTを行います。以下の内容を nvt.mdp に保存します。
define = -DPOSRES
; RUN CONTROL PARAMETERS
integrator = md
dt = 0.002
nsteps = 100000
;
nstxout = 500
nstlog = 500
nstenergy = 500
;
nstlist = 1
pbc = xyz
rlist = 1.0
;
coulombtype = PME
rcoulomb = 1.0
;
vdwtype = cut-off
rvdw = 0.8
dispcorr = enerpres
;
tcoupl = v-rescale
tc_grps = System
tau_t = 0.1
ref_t = 300.0
;
pcoupl = no
;
gen_vel = yes
gen_temp = 300
gen_seed = -1
;
constraints = h-bonds
constraint_algorithm = LINCS
gmx grompp -f nvt.mdp -p pro_metal.top -c em.gro -o nvt.tpr -r em.gro
gmx mdrun -deffnm nvt
NVTはさすがに少し待ちます。AMD Ryzen 5 3600 と GeForce GTX 1050 Ti で以下のようなパフォーマンスが出ました。
Core t (s) Wall t (s) (%)
Time: 256.075 42.681 600.0
(ns/day) (hour/ns)
Performance: 404.869 0.059
計算が無事終わりました。ここで満足したので終わります。
最後に動いている様子を見ておきます。次のように位置を調整しつつ、軽量な軌跡ファイルフォーマットに変換します。
gmx trjconv -f nvt.trr -s nvt.tpr -o nvt.xtc -pbc mol -center
すると次のように、センタリングする対象のグループと出力するグループの2回選択する内容を聞かれます。
Select group for centering
Group 0 ( System) has 2130 elements
Group 1 ( Protein) has 82 elements
Group 2 ( Protein-H) has 40 elements
Group 3 ( C-alpha) has 7 elements
Group 4 ( Backbone) has 24 elements
Group 5 ( MainChain) has 32 elements
Group 6 ( MainChain+Cb) has 39 elements
Group 7 ( MainChain+H) has 46 elements
Group 8 ( SideChain) has 36 elements
Group 9 ( SideChain-H) has 8 elements
Group 10 ( Prot-Masses) has 82 elements
Group 11 ( non-Protein) has 2048 elements
Group 12 ( Other) has 2048 elements
Group 13 ( Ag0) has 2048 elements
Select a group: 1
Selected 1: 'Protein'
Select group for output
Group 0 ( System) has 2130 elements
Group 1 ( Protein) has 82 elements
Group 2 ( Protein-H) has 40 elements
Group 3 ( C-alpha) has 7 elements
Group 4 ( Backbone) has 24 elements
Group 5 ( MainChain) has 32 elements
Group 6 ( MainChain+Cb) has 39 elements
Group 7 ( MainChain+H) has 46 elements
Group 8 ( SideChain) has 36 elements
Group 9 ( SideChain-H) has 8 elements
Group 10 ( Prot-Masses) has 82 elements
Group 11 ( non-Protein) has 2048 elements
Group 12 ( Other) has 2048 elements
Group 13 ( Ag0) has 2048 elements
Select a group: 0
このように Protein を中心にしつつ、系全部を出力するように選んでおきます。
これをVMDで見るとこんな感じになりました。

1フレームは 1 ps に相当します。基本的に表面にぴったりついていて、たまに吸着するサイトが移動しているようです。
まあ上手くできたんじゃないでしょうか。
もっと手短にしたい
なお、金属表面にペプチドを配置するだけならInterfaceのモデルを使わずとも、ASEで十分作れる気がします。
また、Interfaceの力場はLammps用で、LammpsからGROMACSへの変換はInterMolというツールでできますので、Amber経由ではなくLammps経由の方が良いかもしれません。
まあ、Amberのチュートリアルの翻訳ですので。
さらに、いちおうINTERFACE-MDはGROMACS用にも力場ファイルを作っていたようです。
これを使えば、
・ASEで金属表面を作る
・AmberToolsで付着分子の力場を作る(ペプチドならGROMACSでもできるかも)
・GROMACSで金属と分子を合わせたモデルと力場を作り、MDを実行する
とすればできそうです。今回の記事で踏んだ手順よりもうちょっとやりやすくなるのでは?と思っています。
まとめ
Lammpsを覚えるべき。
Discussion