糖タンパク質のMDができるまで
この記事はGROMACSがちょっと使える程度の人間がいろいろやってみた記録をしたものになります。
もし大きな間違いがある、もっと標準的なやり方があるとかがありましたら優しくお教えいただけたら幸いです。
背景
タンパク質のMDというのはいろいろやりがいがあるのですが、チュートリアルに従うと難しいときがあります。
- 小分子ドッキング
- 特殊なアミノ酸残基
- 糖鎖
今回は糖鎖がついたタンパク質をMDで動かしたところまで持っていった話です。
そもそも何が難しいかというと、パラメータぎめです。
古典MDでは原子ごとにLennardJonesポテンシャルなどの分散力のパラメータと点電荷とみなしたときの電荷量を決める必要があります。
原理主義的には目的の分子を2つ並べて配置をいろいろ変えて量子化学計算してフィッティングするのですが、計算が重いこと、それでも溶媒効果などは無くて妥当でないことがあるなど、分子が大きくなると現実的な方法ではありません。
ということで、よくある小分子では、LJポテンシャルパラメータはアルカンの炭素やアルコールの酸素原子みたいに、原子の種類などから典型的なパラメータを辞書的に決めておいて、電荷は量子化学計算から求める方法を取ります。
ですがタンパク質くらいになると量子化学計算もできないので、電荷もデータベース化しておきます。
あとは原子の座標も地味に面倒です。小さい分子だとGaussViewなどでポチポチ作れますが、原子数が数百数千もあるような系はとても手作業ではできず、何かしらのシステム化が要ります。
さて、糖タンパク質の面倒ポイントですが、
- 糖の構成する原子のパラメータ辞書が流行ってない。
- 枝分かれなどもあって構造を表現しにくい。
が主なところかなと思います。これに対して、いろいろなツールがあるのですが、ざっと調べたらCharmmGUI、GlycamWeb、doGlycansがありました。このうちCharmmGUIはライセンスがプライベートな個人に対して使えるのか不明で、GLYCAM-Webは応答が悪いので、ローカルで使えるオープンソースのdoGlycansを使ってみました。
うまくいかなかったもの
AmberTools のライブラリ内に leaprc.GLYCAM_06j-1 とかあって、標準的な糖タンパク質なら何か別ツールに頼らなくてもAmberToolsで完結できるかな、と思いましたが、データベースからダウンロードしてきたPDBの残基名・原子タイプ名だとうまく認識されないのか、あるいは何か手順を踏めていないのか、うまくいきませんでした。
doGlycansは内部で持っている各種データは結局AmberToolsから拾ってきてちょっと足しているようなので、AmberTools内でカバーできるものならできる気がするのですが。
もし可能であれば 「Gromacs用パラメータができるまで」の「doGlycans」の節が全てAmberToolsというかtLeapの処理に変わります。もしやり方が分かれば追記します。
ただ、 tleap ができるとしたら用意したpdbファイルに糖鎖がちゃんとあるときで、「ダウンロードしたpdbにはないけど何らかの事情で残基にこの糖鎖を付けたシミュレーションをしたい」「糖鎖の種類を変えたシミュレーションをしたい」とかでは doglycans も便利だと思いますので、やり方を書いて残しておきます。
pdbに付いていない糖を付けるには、tleapによるポリマーの構築と似た手順な気がするのですが、そうなると糖単体のacとprepファイルを用意しないといけない気がする。
追記
端的に言いますと、RCSBでダウンロードしたPDBファイルにある糖鎖は、残基名のほか原子名も変えないといけませんでした。正しい残基名、原子名はGlycoShapeから得られます。
当該糖鎖のページを表示、ファイルをダウンロードすると、そのzipファイル内にGLYCAM向けのpdbファイルをがあります。これにGLYCAM向けの残基名、原子名がありました。VMDで両方の残基名・原子名を表示させ、対応する原子を探して修正するものを探します。

今回、アセチルグルコサミンだと以下の原子の名前を変えるとよさそうでした。残基名は 4YB または 0YB です。
| RCSB | GLYCAM |
|---|---|
| H81 | H1M |
| H82 | H2M |
| H3M | H83 |
| CME | C8 |
| C7 | C2N |
| O7 | O2N |
| HN2 | H2N |
| HO3 | H3O |
| H4O | HO4 |
| H6O | HO6 |
まあ世の中にはこういう対応表だったり、何なら自動で変換してくれるツールを出している人が要るかもしれません。ただ今回は糖が2つだけだったので、glycoshapeからダウンロードしたpdbを参考にして手でやってしまい、テストをしました。自動化を考えるのは上手くいってからでよいだろうと。
で、こうやって糖鎖の残基名、原子名のほか、ジスルフィド結合などMD向けに処理したpdbファイルを protein.pdb とします。以下のような感じでとりあえずAmberのパラメータは作れそうです。
source leaprc.GLYCAM_06j-1
source leaprc.gaff2
source leaprc.protein.ff14SB
prot = loadpdb protein.pdb
bond prot.78.C1 prot.18.ND2 # 4YB-ASN のN-グリコシル結合
check prot
# 水溶液とかイオン追加するならここで
saveamberparm prot system.prmtop system.inpcrd
quit
この糖タンパク質は GlcNAc-GlcNAc-ASN(18) という糖鎖を持っている状況です。このうち糖―タンパク質間の結合はあらわに指定する必要がありました。
注意:たぶんこれは不完全です。なんか糖を付けた18番目の残基のND2に、糖が付くべき結合に水素も追加されてしまっていて、ND2が4価の窒素になっています。
また、テストとしてこの記事のこの後で出すタンパク質 1CDS についてやってみたのですが、電荷も -1.8程度となり、なにか原子の過不足が起こっている感じがします。
で、これでできたAmber用パラメータを変換しようとしました。
import parmed as pmd
mol = pmd.load_file("system.prmtop", "system.inpcrd")
mol.save("system.top", format="gromacs")
mol.save("system.gro")
すると以下のようなエラーが出ます。
parmed.exceptions.GromacsError: Structure has mixed 1-4 scaling which is not supported by Gromacs
これを調べると、GROMACSでは対応していない機能だということです。
これ以上についてはAmberを深く知らないといけないようです。ということでこの方向での検討はここまで。
必要なもの
OS
WSL2などLinux環境があるとよいです。必要に応じてパッケージをインストールできるとなおよいです。
Gromacs
普通にインストールします。
AmberTools
ないなら無いで何とかなりますが、あると便利なので。
doGlycans
これはpipでインストールします。
URL: https://bitbucket.org/biophys-uh/doglycans/src/main/
cd /path/to/doglycans/
python -m pip install .
cd doglycans
mv doglycans.py main.py
最後の doglycans.py の名前を変えるのは、パッケージ内にパッケージと同名ファイルがあると
ModuleNotFoundError: No module named 'doglycans.prepreader'; 'doglycans' is not a package
ていうエラーが出るためです。 doglycans.py には特にライブラリとして呼び出されることのない実行スクリプトファイル的なものでしたので、適当な名前に変えました。
Gromacs用パラメータができるまで
ダウンロードと処理
適当な作業ディレクトリを workdir とします。この中に前処理、MD実行も合わせると大量のファイルができますので、処理内容のめどがついたら適宜フォルダ分けすると良いと思います。
cd /path/to/workdir
wget https://files.rcsb.org/download/1CDS.pdb
これはどうもNAG2つが連なった糖鎖が1つだけついている小さめのたんぱく質CD59について溶液NMRにより10くらいのコンフォーマーを推定したもののようです。それらが1つのPDBファイルに入っています。
まず、1つ目の構造だけとってきつつ、水素を除くのと糖鎖などHETATMを除く処理を行います。手でやってもいいのですが、AmberToolsがあるとコマンド一つでできます。
pdb4amber -p -y --model 1 1CDS.pdb > 1CDS_amber.pdb
あと10個あるシステイン残基はすべてSS結合していますので、それ用の残基名に変えておきます。
sed -i s/CYS/CYX/g 1CDS_amber.pdb
doGlycans
ここからはdoGlycanのドキュメント( bitbucketリポジトリのpdfファイル )の例に沿った処理を行います。
水素化と原子タイプ識別処理を行います。
gmx pdb2gmx -f 1CDS_amber.pdb -o ProtH.pdb -p ProtH.top
今回はAmber力場、TIP3Pということで 1 <Enter> 1 <Enter> と入力します。続いて生成した ProtH.top を ProtH.itp に変えつつ、分子定義以外のところをコメントアウト・削除します。コメントアウト対象は [ moleculetype ]に関するところ以外、つまり #include や [ system ] などです。手作業でもいいですが、この時は以下のコマンドでできました。
grep -v "#" ProtH.top | head -n -17 > ProtH.itp
つづいて、糖鎖の定義です。doglycansにより、原子タイプを上手く設定しつつ糖鎖の原子を配置できます。 doGlycans に読み込んでもらうファイルを作成します。
A=P.18/HD22:-(ND2,C1,<Ae>)-4YB-(O4,C1,<a>)-4YB
Ae=ND2,C1,[CG ND2 C1 C2 156.9]
a = O4,C1,[C3 C4 O4 C1 134.0 C4 O4 C1 C2 166.3]
文法は doglycansのドキュメント( bitbucketリポジトリのpdfファイル )を参照してください。ざっくりいうと、18番目の残基にある HD22 水素を削除して ND2 窒素に n-β-アセチルグルコサミン(4YB、 4 は結合箇所、 Y はグルコサミン、 B はβを示す )の C1 原子をつなげ、さらに 1つめの 4YB の O4 原子の二つ目の 4YB の C1 原子をつなげています。
Ae とか a は2面角の定義です。最初の A はたぶん鎖のラベルかな?と思いつつ適当にしてみて、うまくいったのでそのままですが、よくわかっていません。
なお、 156.9 とか 134.0 とかの数字は結合の2面角で、適当です。実はこのたんぱく質をやる前に別の糖タンパク質でも試していて、そのときはAvogadroで頑張って拾ってきたのですが、なんか配置は再現しませんでした。二面角の選択原子の順番を間違えたか、AvogadroとdoGlycansで角度の基準が違うのかもしれません。
この sequence_file.seq ファイルを用意したのち、糖鎖をつなげるコマンドを実行します。
python /path/to/doglycans/doglycans/main.py -f /path/to/doglycans/dat/glycam06h.itp -p /path/to/doglycans/dat/GLYCAM_06h.prep -s sequence_file.seq -c ProtH.pdb -i ProtH.itp -o ProtG.pdb -t ProtG.itp -B --amber
これで ProtG.pdb と ProtG.itp が生成されます。ただこの doglycans のスクリプト、一部処理をさぼっていて、たとえば電荷の調整がされていません。
まず、糖鎖をつなげた ASN の窒素原子について、水素原子が炭素原子に変わったため、水素原子の電荷が消えます。そのため、とりあえず窒素原子に水素原子の電荷を集約しておきます。
253 N 18 ASN ND2 253 -0.4267 14.007
ついでに、GROMACSのMDでは良くあることなのですが、合計の電荷が0.001程度ずれていました。仕方ないので最初の原子に押し付けます。どれだけズレるかは系によります。
; Residue 1 LEU q=1.000 -> reduce 0.002101
1 N3 1 LEU N 1 -0.626801 14.007 ; qtot -0.6247
この記事を参考にして別の系や別の計算機で計算した場合、修正するべき値も違うと思われます。修正値は後々になって分かります(gmx grompp コマンドを打った場合など)。そのあとに改めて修正して大丈夫です。
あと、 doglycans のExampleからファイルをとってきます。
cp /path/to/doglycans/Examples/EGFR/AMBERff/top/glycam06h.itp .
cp /path/to/doglycans/Examples/EGFR/AMBERff/top/ions_Dang.itp .
cp /path/to/doglycans/Examples/EGFR/AMBERff/top/ions_nb_Dang.itp .
そして、このあとの手順で gmx genions でイオンを追加する段階があるのですが、その原子名がこのコピーしてきたファイルに合わせるのが面倒ですので、このファイルの方を修正します。具体的には moleculetype の名前の末尾の _d 削除します。たとえば Na_d から NA にします。
[ moleculetype ]
; Name nrexcl
CL 1
(中略)
[ moleculetype ]
; Name nrexcl
NA 1
最後に system.top を用意します。
#include "amber03.ff/forcefield.itp"
#include "glycam06h.itp"
#include "ions_nb_Dang.itp"
#include "ions_Dang.itp"
#include "ProtG.itp"
#include "amber03.ff/tip3p.itp"
[ system ]
; Name
[ molecules ]
; Compound #mols
Molecule 1
これで糖タンパク質の座標pdbとパラメータが定義されたファイルができました。
Gromacsによる前処理
まず水に溶かします。
gmx solvate -cp ProtG.pdb -p system.top -box 7
これにより system.top に SOL 分子が追加されつつ、 out.gro ができます。
続いてイオン化をするのですが、これには tpr が要求されますので、罪のない mdp ファイルを用意します。
integrator = steep
nsteps = 5000
nstlog = 1
emstep = 0.0005
dt = 0.0002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = no
Lincs-iter = 2
Lincs-order = 4
nstlist = 1
pbc = xyz
rlist = 1.0
coulombtype = Cut-off
rcoulomb = 1.0
rvdw = 1.0
cutoff-scheme = Verlet
まあまだ削れる気はします。それはともかく、これでいったん grompp をします。
gmx grompp -f ionize.mdp -c out.gro -p system.top -o ionize.tpr
ここで出力の Warning を見ていると系の合計電荷が分かりまして、それを基にして上の方でやっていた 0.002 程度の電荷の調整を行いました。続いて中性になるように水分子をいくつか Na に置換します。
gmx genion -s ionize.tpr -o ionized.gro -p system.top -pname NA -nname CL -neutral
ここでどのグループの分子を置換するか聞かれますが、 15 (SOL)を選択します。
これでたんぱく質に糖鎖が付き、水に溶け、中性になるようイオンが追加された ionized.gro と、その力場ファイル system.top ができました。
お疲れ様です。あとは流れで大丈夫です。
MD実行
ここからは実はあまりdoglycansの使い方に関係ないので、普通にGROMACSのMDを実行しましたということを残しておきます。
エネルギー最小化
integrator = steep
nsteps = 5000
nstlog = 1
emstep = 0.0005
dt = 0.0002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = no
Lincs-iter = 2
Lincs-order = 4
nstlist = 1
ns_type = grid
pbc = xyz
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
cutoff-scheme = Verlet
gmx grompp -c ionized.gro -f min.mdp -p system.top -o em.tpr
gmx mdrun -deffnm em
ちょっと分子の様子を見てみます。

分かりにくいので以下のように修正。
gmx trjconv -f em.gro -s em.tpr -pbc mol -center -o em.pdb
何を中心に据えるか、どの原子を出力するかを聞かれますので、 1 <Enter> 0 <Enter> と入力。生成された em.pdb を見てみます。

よさそうですね。見やすくしてみます。
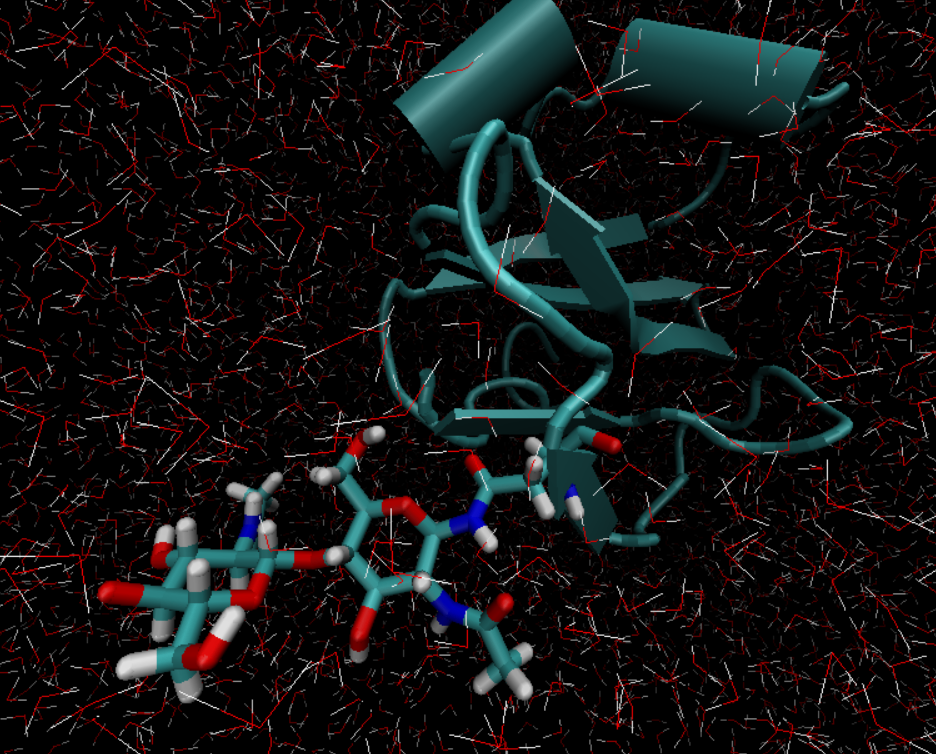
このとき、 Licorice で表現している残基の Selection は residue 17 or resname 4YB です。
平衡化
最初は系に対して急激な変化がありそうなので、熱浴を弱めにしたNPTののち強くして2回目のNPTという形にしました。
1回目NPT
integrator = md
nsteps = 5000
nstxout = 500
dt = 0.002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = no
Lincs-iter = 1
Lincs-order = 4
cutoff-scheme = Verlet
nstlog = 1
nstlist = 10
ns_type = grid
pbc = xyz
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
tcoupl = V-rescale
tc-grps = System
tau-t = 1.0
ref-t = 300
pcoupl = c-rescale
;pcoupl = Parrinello-Rahman
;tau-p = 1.0
tau-p = 10.0
ref-p = 1.0
pcoupltype = isotropic
compressibility = 4.5e-5
gen-vel = yes
gen-temp = 300
gmx grompp -c em.gro -f npt1.mdp -p system.top -o npt1.tpr
gmx mdrun -deffnm npt1
2回目NPT
integrator = md
nsteps = 10000000 ; 20 ns
nstxout = 5000
dt = 0.002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = yes
Lincs-iter = 1
Lincs-order = 4
cutoff-scheme = Verlet
nstlog = 500
nstlist = 10
ns_type = grid
pbc = xyz
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
tcoupl = V-rescale
tc-grps = System
tau-t = 1.0
ref-t = 300
pcoupl = c-rescale
tau-p = 1.0
ref-p = 1.0
pcoupltype = isotropic
compressibility = 4.5e-5
gen-vel = no
gmx grompp -c npt1.gro -f npt2.mdp -p system.top -o npt2.tpr
gmx mdrun -deffnm npt2
今回の系は 33686 原子あるのですが、 core-i7 14700F、 RTX 5060 Ti で 400 ns/day ちょっとでした。
On 1 MPI rank, each using 14 OpenMP threads
Activity: Num Num Call Wall time Giga-Cycles
Ranks Threads Count (s) total sum %
--------------------------------------------------------------------------------
Neighbor search 1 14 100001 143.752 4249.669 3.4
Launch PP GPU ops. 1 14 19900001 1030.785 30472.515 24.4
Force 1 14 10000001 1228.495 36317.307 29.0
PME GPU mesh 1 14 10000001 742.364 21946.102 17.5
Wait GPU NB local 1 14 10000001 21.839 645.627 0.5
Wait GPU state copy 1 14 20500003 979.467 28955.445 23.2
NB X/F buffer ops. 1 14 100001 2.924 86.434 0.1
Write traj. 1 14 2005 6.779 200.412 0.2
GPU constr. setup 1 14 1 0.000 0.012 0.0
Kinetic energy 1 14 200001 7.440 219.942 0.2
Rest 66.242 1958.281 1.6
--------------------------------------------------------------------------------
Total 4230.088 125051.745 100.0
--------------------------------------------------------------------------------
Breakdown of PME mesh activities
--------------------------------------------------------------------------------
Wait PME GPU gather 1 14 10000001 6.104 180.460 0.1
Reduce GPU PME F 1 14 10000001 2.675 79.072 0.1
Launch PME GPU ops. 1 14 90000009 718.404 21237.770 17.0
--------------------------------------------------------------------------------
Core t (s) Wall t (s) (%)
Time: 59221.148 4230.088 1400.0
1h10:30
(ns/day) (hour/ns)
Performance: 408.502 0.059
パラメータの妥当性はともかくとして、MD自体は問題なく動いているようです。
ちなみ入力ファイルを別に作って細かく軌跡を出すようにしてみた結果の動画をツイートしています。
Discussion