膜たんぱくMDチュートリアル 続き
せっかくなので、チュートリアルになかった量でも計算してみます。
動径分布関数
脂質分子同士の分布を見ようと思います。
ちょっと考えたこと
GROMACSには gmx rdf というコマンドがあり、これを使うことにします。
ただし問題があります。
2次元の膜を形成する分子の分布を知りたいときは、同じ膜内の分子を想定し、かつ膜が存在する2次元空間だけで考えたいところです。
層が異なっていてxy座標が同じ分子があるとほぼ0の距離に存在してしまうことになります。
オプションで -xy はあり、これで2次元での計算は可能なのですが、層を分けるのはやってくれません。
層の分離は自分でグループ分けします。
グループ分け
まず適当なgroファイルを開いてみます。そして OL が脂質分子についているラベルですが、この原子のZ座標(5列目)をみて、急に大きくなる原子を探します。
1OL H17S18760 7.516 7.787 4.133 0.3406 -0.9108 -0.2837
1OL C11818761 7.480 7.958 4.002 -0.7066 -0.4763 0.0594
1OL H18R18762 7.454 8.025 4.084 -0.9015 2.4436 -2.2260
1OL H18S18763 7.533 8.015 3.925 -1.0912 1.2106 1.0073
1OL H18T18764 7.382 7.928 3.965 -1.6551 1.6909 0.6416
2PGR C1118765 7.902 7.514 2.550 0.4588 -0.2121 -0.3348
2PGR O1218766 7.948 7.412 2.512 0.4467 -0.8948 -0.4475
2PGR O1118767 7.859 7.600 2.453 0.5015 0.7064 -0.3239
2PGR C118768 7.909 7.594 2.317 0.4825 -0.1473 -0.3351
2PGR HR18769 7.921 7.498 2.266 -2.2045 -1.3375 1.0802
2PGR HS18770 7.830 7.642 2.260 -0.8818 -1.0249 0.7341
2PGR C218771 8.029 7.691 2.300 -0.5333 -0.3500 0.0301
2PGR HX18772 8.061 7.687 2.195 -0.8739 -0.9375 -0.0588
2PGR C318773 8.149 7.660 2.392 0.0707 -0.5667 0.2294
・・・
3OL H17R18840 7.925 8.172 3.809 -0.5526 1.5218 2.4311
3OL H17S18841 8.024 8.067 3.912 0.0213 -2.2338 -0.7593
3OL C11818842 7.817 8.124 3.983 -1.1535 -0.1491 0.2764
3OL H18R18843 7.718 8.128 3.938 -1.0979 -0.3315 0.1325
3OL H18S18844 7.843 8.223 4.020 0.5585 -1.1887 2.0677
3OL H18T18845 7.808 8.056 4.067 -2.1910 -0.4326 -0.0535
1OL C1218846 5.745 7.019 5.534 -0.1490 -0.1248 -0.3564
1OL H2R18847 5.812 7.094 5.492 1.4087 -0.6016 1.1680
1OL H2S18848 5.786 6.978 5.626 0.9455 -0.4031 -0.9579
1OL C1318849 5.738 6.897 5.444 -0.7586 0.4836 0.3874
1OL H3R18850 5.654 6.833 5.472 -0.8598 0.6254 0.4118
1OL H3S18851 5.828 6.837 5.461 -1.2270 -1.3719 -2.8577
・・・
H18T18845 まではZ座標がせいぜい 4 以下だったのが、C1218846 からは急に5を超えてきました。ここで層が変わったものとします。
また、脂質分子は PE PGR OL の3種類のラベルがあります。
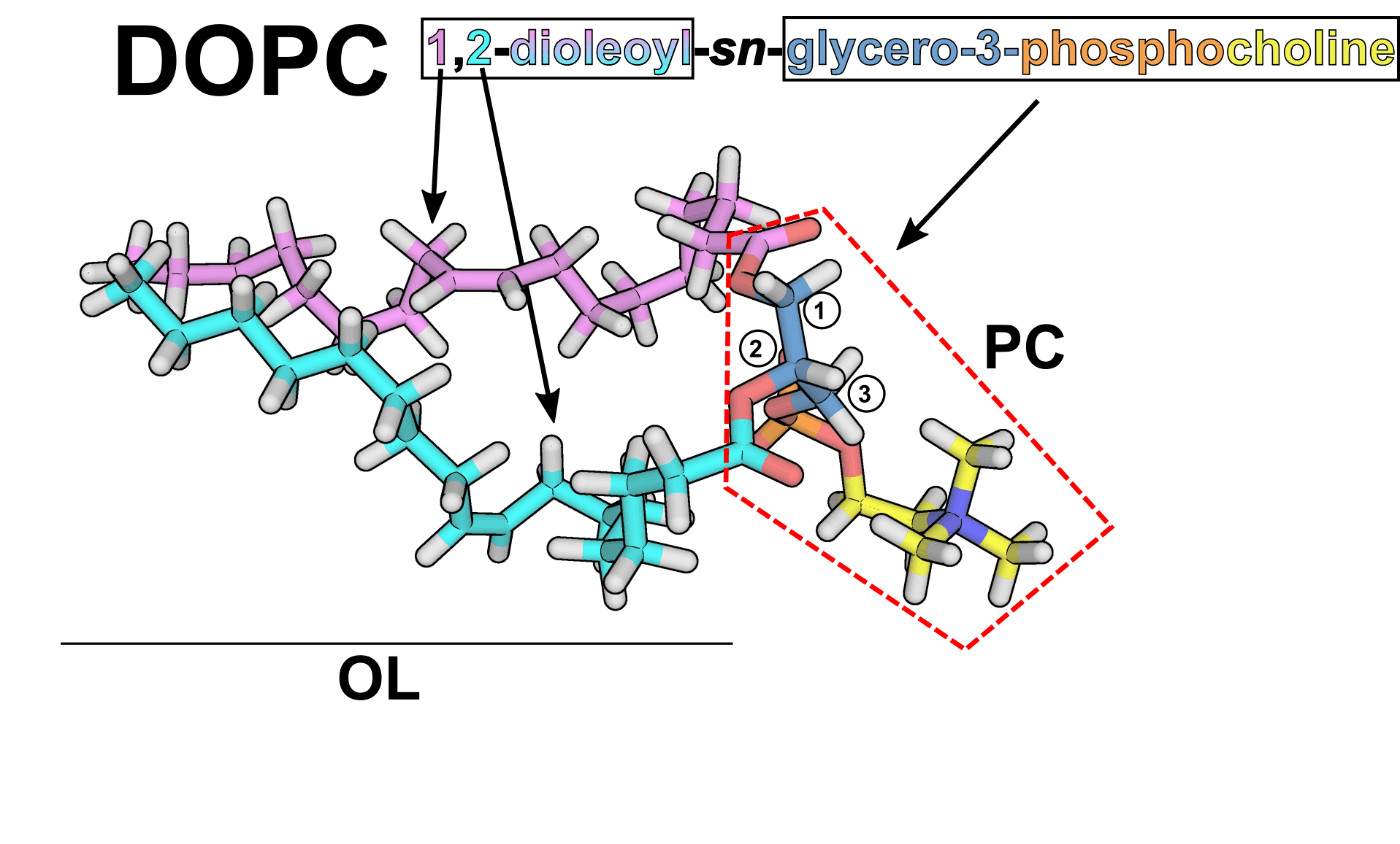
図は https://ambermd.org/tutorials/advanced/tutorial38/index.php から引用しているもので、ちょっと違う脂質分子ですが、
PC が今回のシミュレーションで使っている2種類の脂質の PE PGR にあたる部分、OL は足の部分です。
MDを流していると、足の部分は結構絡み合うことがわかります。するとOL まで含めた分子の重心の距離はほぼ0になったりします。
そこで、 PE PGR のラベルのついた原子をまとめてしまい、18845 番目の原子を境として、2つのグループに分けます。
GROMACSではこういうグループを index.ndx などのファイルで管理でき、これを作成するコマンドが gmx make_ndx です。これを実行すると対話処理が始まりますので、以下のように実行します。
あ、前回のシミュレーション から npt3.gro を持ってきます。
gmx make_ndx -f npt3.gro
> a 1-18845 & r PE PGR
Found 18845 atoms in range 1-18845
Found 5635 atoms with residue names PE PGR
Merged two groups with AND: 18845 5635 -> 2950
24 a_1-18845_&_PE_PGR : 2950 atoms
> name 24 lower_oilbase
> a 18846-63465 & r PE PGR
Found 44620 atoms in range 18846-63465
Found 5635 atoms with residue names PE PGR
Merged two groups with AND: 44620 5635 -> 2685
25 a_18846-63465_&_PE_PGR: 2685 atoms
> name 25 upper_oilbase
> q
これで index.ndx ができました。
lower_oilbase には Z 座標が 4以下の油が、upper_oilbase には 4以上の油が含まれているかと思います。
動径分布関数
これを使って動径分布関数は以下のように計算します。
gmx rdf -f prod.trr -s prod.tpr -n index.ndx -xy -selrpos whole_res_com -seltype whole_res_com -ref upper_oilbase -sel upper_oilbase
または
gmx rdf -f prod.trr -s prod.tpr -n index.ndx -xy -selrpos whole_res_com -seltype whole_res_com -ref lower_oilbase -sel lower_oilbase
のどちらかを実行すると、 rdf.xvg ができます。
-ref と -sel に同じグループを指定していることで自分自身の動径分布関数を求めています。
selrpos と seltype に whole_res_com を指定し重心間距離としました。これのオプションはすごいいっぱいありますけど、これはとても大きいたんぱく質の一部の残基とかを考えたいとき用なので、今回のような小さい分子の時は com が入っていれば大丈夫かと思います。
rdf.xvg は xmgrace で表示できます。
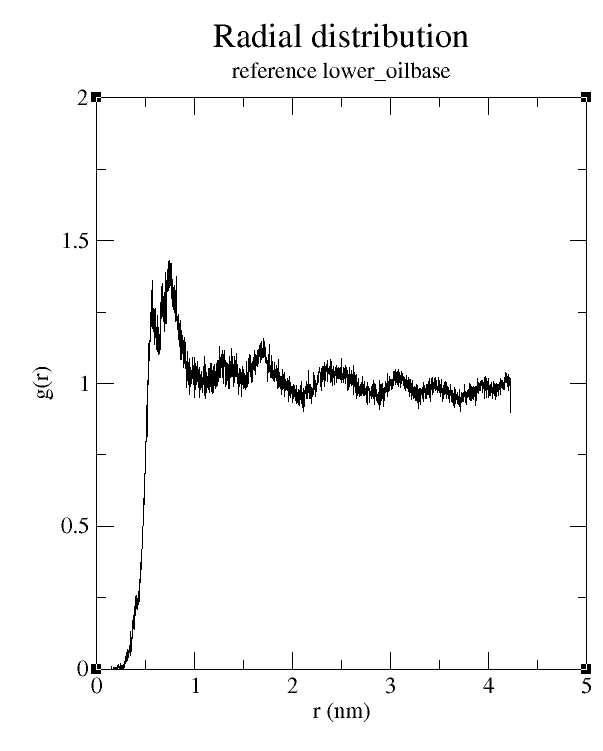
残念ながらウチはこの話は詳しくないのでこの結果が妥当かどうかがわかりません。
Umbrella Sampling
カリウムイオンチャネルたんぱく質中をカリウムイオンが通過するときに感じる平均力ポテンシャル表面(Potential of Mean Force、PMF)を、Umbrella Samplingという手法を使って計算してみます。
今回は込み入った設定を行いますので、Umbrella Samplingが初めての方はもっと簡単なUmbrella Sampling自体のチュートリアルを別に行うことをお勧めします。
たとえばこれ:http://www.mdtutorials.com/gmx/umbrella/index.html
座標設定
たんぱく質とカリウムイオンの距離を
くというのが今回の目標です。
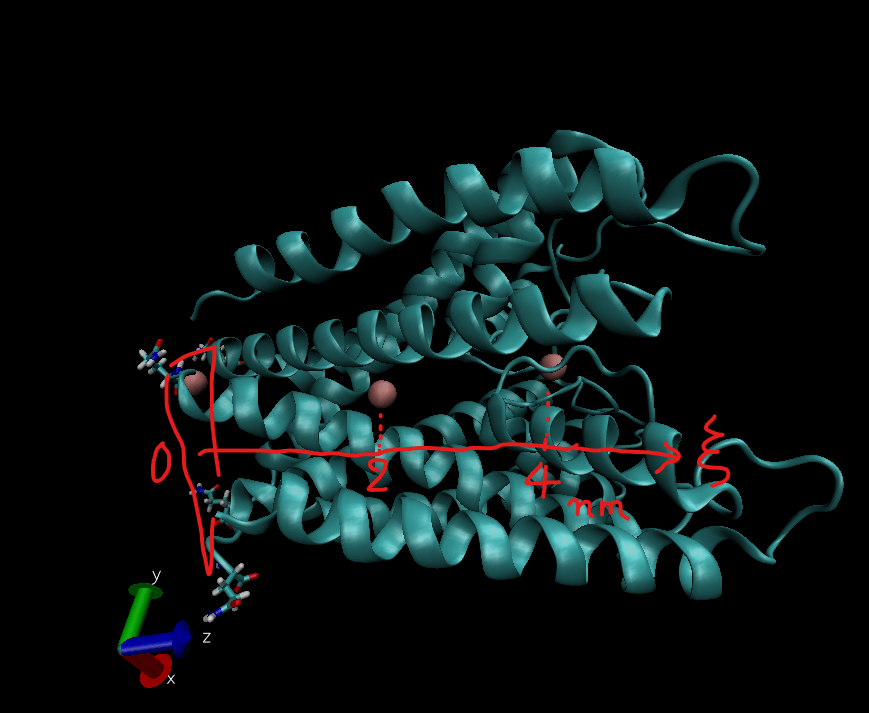
この図のように、左側(おそらく膜内側)の端にある4つの残基の座標を
座標の基準にした残基の根拠ですが、GROMACSでpull機能を使う時、シミュレーションの箱のサイズの半分より大きい分子、つまりたんぱく質の重心を指定することができませんでした。
そこでたんぱく質とカリウムイオンの距離を指定するにあたり、適当な残基中の原子または残基の重心を基準座標とすることにしました。
今回は単に OXT タイプの原子を持っていてファイル中で特定しやすかったというだけです。もっといい基準があると思います。
今回の計算には人工的なばねのような力を与えるpull機能 (Non-equilibrium pulling)を使います。
グループ間の距離を評価し、それに対し設定したばね定数に基づいてグループ中の原子に力をかけます。
しかし、この距離は必ず正となります。基準の残基の重心とカリウムイオンのZ座標がほぼ一致しているとき、0を中心として振動すると考えられますが、この距離に基づいて座標を決めてしまうと0付近の正の値の座標でサンプリングが過剰に行われてしまいます。
負の値も取りうる
安直な発想としては系の原点とカリウムイオンの距離(絶対座標)をつかい、これにばねを設定することですが、こうすると無限の質量をもつ系とカリウムイオン、カリウムイオンとたんぱく質という相互作用により、たんぱく質が並進してしまう危険性があります。
なので、絶対座標を基準としつつ、並進を打ち消すような座標を使おうと思います。
絶対座標で動かないある点を基準点にして、基準点からカリウムとの距離と、基準点からたんぱく質の距離を考え、両者の差を取ります。
基準点の座標がキャンセルされて絶対座標と原子間に力が掛からないので並進運動が生じないだろうという発想です。(座標とはどう定義されるかを考えれば自然な発想ではと思います)
まず、カリウムのZ座標を
また、基準となる座標を
必ず
カリウムと基準点の距離は
周期境界条件
系のZ軸方向のサイズを
しかし距離(Z軸上の差)となると、イメージセルを適切に選べば必ず
そこでGROMACSは
そして時間発展の結果
今回の系では
このとき
お断り
すみません、実はこの座標の定義で本当に並進運動が排除できているかは確認していません。
もし問題がありましたら、この章についてはpull機能における pull-coord-geometry = transformation のサンプルとして参考にする程度にはできると思います。
あと結果についても、並進運動が大きくなる前にサンプリングMDを終えているかと思いますのでそこまで的外れというわけでもないのではと思います。
どうしても
準備
以上が計算の背景となります。これから実際の作業に入ります。
たんぱく質1BL8には内部にカリウムイオンを3つ含んでいます。
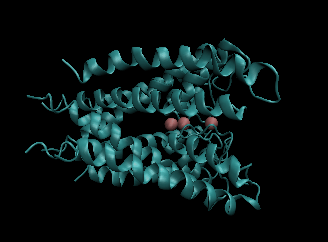
このまま動かすとカリウムイオン同士の反発が考えられるので、スキャンのため動かす1個をのこし、2個を外に放り出します。
あと前回のチュートリアルから npt3.gro を持ってきます。
グループ分け
GROMACSでは人工的な力を与えることができるのですが、どの原子に力を与えるかの指定に index.ndx により指定されるグループを用います。
カリウムイオンは 5893 ~ 5895 番目の原子なのですが、 5895 番目の原子をスキャンに用い、それ以外を外に出します。
よってスキャン用のカリウムイオンと外に出すための2個のイオン、座標の基準となる残基グループの3つのグループを作ります。
gmx make_ndx -f npt3.gro
> a 1-5894 & r K
> name 24 K_out
> a 5895
> name 25 K_target
> a 1456-1473 | a 2929-2946 | a 4402-4419 | a 5875-5892
> name 26 protein_base
> q
これで index.ndx ファイルができたかと思います。
イオン排出
GROMACSでは、 pull により人工的な力を与えることができます。
まず、以下のように kickout.mdp を作ります。
integrator = sd
nsteps = 1000
nstxout = 10
dt = 0.002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = no
Lincs-iter = 1
Lincs-order = 4
cutoff-scheme = Verlet
nstlog = 10000
nstlist = 10
pbc = xyz
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
tc-grps = System
tau-t = 1.0
ref-t = 300
pull = yes
pull-ngroups = 1
pull-ncoords = 1
pull-group1-name = K_out
pull-coord1-groups = 0 1
pull-coord1-type = constant-force
pull-coord1-geometry = direction-periodic
pull-coord1-vec = 0.0 0.0 -1.0
pull-coord1-k = 2000
やっていることは K_out に属するカリウムイオン2つをZ軸の負の方向に力をかけているだけです。この計算ではまだ
gmx grompp -c npt3.gro -p system.top -f kickout.mdp -o kickout.tpr -n index.ndx --maxwarn 1
gmx mdrun -deffnm kickout
pull-coordN-groups に 0 つまり絶対座標があると、並進運動が生じる恐れがあり、そのため結果にそれに起因した構造がでたりMDが正しくなくなる可能性があるので、grompp の時にWarningが出ます。
あと、圧力に関する熱浴を外しています。これは pull-coord1-geometry = direction-periodic の要求によるものです。
今回はそれを承知の上でやっているので、 --maxwarn 1 オプションを追加しました。
これでカリウムイオンが2つ外に出ます。
(もしかしたら gro ファイル中の座標の数値を直接書き換えてもよかったかもしれない)
平衡化とサンプリング
mdpファイル
Umbrella Samplingでやることは、サンプルを取りたい
まずは平衡化用のインプットファイルは以下のようになります。
integrator = md-vv
nsteps = 500000
nstxout = 50000
dt = 0.002
constraints = h-bonds
constraint-algorithm = Lincs
continuation = no
Lincs-iter = 1
Lincs-order = 4
cutoff-scheme = Verlet
nstlog = 10000
nstlist = 10
pbc = xyz
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
tcoupl = V-rescale
tc-grps = System
tau-t = 1.0
ref-t = 300
pcoupl = Parrinello-Rahman
tau-p = 1.0
ref-p = 1.0 1.0
pcoupltype = semiisotropic
compressibility = 4.5e-5 4.5e-5
pull = yes
pull-ncoords = 3 ; x1, x2, ξ の3つの座標がある
pull-ngroups = 2
pull-group1-name = K_target
pull-group2-name = protein_base
; カリウムイオンの座標 x1 = z1 - z0 用の定義
pull-coord1-type = umbrella
pull-coord1-geometry = distance
pull-coord1-groups = 0 1
; z0 に相当、座標に応じてここを変える
pull-coord1-origin = 0.0 0.0 0.0
pull-coord1-dim = N N Y
pull-coord1-k = 0
; たんぱく質の座標 x2 = z2 - z0 用の定義
pull-coord2-type = umbrella
pull-coord2-geometry = distance
pull-coord2-groups = 0 2
; z0 に相当、座標に応じてここを変える
pull-coord2-origin = 0.0 0.0 0.0
pull-coord2-dim = N N Y
pull-coord2-k = 0
; x1 と x2 の間のばねの定義
pull-coord3-type = umbrella
pull-coord3-geometry = transformation
pull-coord3-expression = x1 - x2
pull-coord3-dim = N N Y
; サンプリング場所に応じて以下2つを変える
pull-coord3-k = 400
pull-coord3-init = 5.0
pull-nstxout = 50
pull-nstfout = 0
次にサンプリング用のファイル template_prod.mdp ですが、
今回は平衡化する時間とサンプリング時間を同じにしていたので、インプットファイルの前半は同じです。
さらに、pull 関連もほとんど同じです。
Umbrella Samplingでは XXXX_pullx.xvg を使います。これを制御するのは pull-nstxout で、これもデフォルトの 50 ステップでよければ2つのテンプレートからこの pull-nstxout とか pull-nstfout を削除してしまってもいいでしょう。このとき両者のテンプレートファイルは完全に同じです。
今回はもう同じものとしてしまいます。
cp template_eq.mdp template_prod.mdp
実行
pull_coord3_init を XX に設定します。このファイルを eq_XX.mdp prod_XX.mdp とします。
gmx grompp -c kickout.gro -p system.top -f eq_XX.mdp -o eq_XX.tpr -n index.ndx --maxwarn 1
gmx mdrun -deffnm eq_XX
gmx grompp -c eq_XX.gro -p system.top -f prod_XX.mdp -o prod_XX.tpr -n index.ndx --maxwarn 1
gmx mdrun -deffnm prod_XX
grompp でWarningが出ます。
シェルスクリプト
まず、bashで pull_coord3_init にセットしたい値を第一引数に、 pull_coord3_k を特別に決めたい値があれば第二引数にとる関数を作ります。
function process_i(){
distance=$1
k_const=$2
eq_job=eq_${distance}
prod_job=prod_${distance}
# Bashは整数だとifとかで使いやすいので、ピコmに変換したものを用意
distance_pm=$(awk "BEGIN {x=${distance}*1000; print (x>0)? int(x + 0.5): int(x - 0.5);}")
# z_0 を カリウムイオンの設定したい値に応じて変更
z_origin=$(awk "BEGIN {x=${distance}; print (x>0)? ((x >= 4)? 3.2: 1.6): 0.8;}")
# 第2引数が無い場合、 ξ< 0 なら400を、 0 < ξ < 3 なら 1500を、3以上なら 1000 を設定
if [ "k$k_const" = "k" ]; then
k_const=400
if [ "$distance_pm" -ge "0" -a "$distance_pm" -lt "3000" ]; then
k_const=1500
fi
if [ "$distance_pm" -ge "3000" -a "$distance_pm" -lt "6000" ]; then
k_const=1000
fi
fi
if [ ! -f ${eq_job}.gro ]; then
# まずはパラメータを修正
sed "s/pull-coord3-init = 5.0/pull-coord3-init = ${distance}/" template_eq.mdp > ${eq_job}.mdp
sed -i "s/pull-coord3-k = 400/pull-coord3-k = ${k_const}/" ${eq_job}.mdp
sed -i -r "s/(pull-coord[12]-origin) = 0.0 0.0 0.0/\1 = 0.0 0.0 ${z_origin}/g" ${eq_job}.mdp
# 原点の位置によってはx1 の符号が反転する
if [ "$z_origin" = "3.2" ]; then
sed -i -r "s/(pull-coord3-expression) = x1 - x2/\1 = x1 + x2/g" ${eq_job}.mdp
fi
# 平衡化MD 実行
gmx grompp -c kickout.gro -p system.top -f ${eq_job}.mdp -o ${eq_job}.tpr -n index.ndx --maxwarn 1
gmx mdrun -deffnm ${eq_job}
fi
if [ ! -f ${prod_job}.gro ]; then
# まずはパラメータを修正
sed "s/pull-coord3-init = 5.0/pull-coord3-init = ${distance}/" template_prod.mdp > ${prod_job}.mdp
sed -i "s/pull-coord3-k = 400/pull-coord3-k = ${k_const}/" ${prod_job}.mdp
sed -i -r "s/(pull-coord[12]-origin) = 0.0 0.0 0.0/\1 = 0.0 0.0 ${z_origin}/g" ${prod_job}.mdp
if [ "$z_origin" = "3.2" ]; then
sed -i -r "s/(pull-coord3-expression) = x1 - x2/\1 = x1 + x2/g" ${prod_job}.mdp
fi
# サンプリング MD 実行
gmx grompp -c ${eq_job}.gro -p system.top -f ${prod_job}.mdp -o ${prod_job}.tpr -n index.ndx --maxwarn 1
gmx mdrun -deffnm ${prod_job}
fi
}
あとはこの計算セットをサンプリングする場所すべてで繰り返すだけです。
gro ファイルができてるかどうかで計算終了しているかどうかを判定しています。もしあればその計算をスキップします。
とりあえず
for i in `seq 32`
do
distance=$(awk "BEGIN {print ($i - 4)*0.2}")
process_i $distance
done
さらに試行錯誤しているうちにプラスαでサンプリングが必要なところを追加でやっておきます。
場所によってはたんぱく質からかかる力が強く弾き飛ばされてしまうため、 pull-coord3-k を大きくする必要がありました。だから process_i は必要があれば第二引数を取るようにしました。
process_i 1.1
process_i 1.28 2500
process_i 1.99 3000
AWH(accelerated weight histogram)
最後に、大量にできた prod_XX_pullx.xvg を使って自由エネルギー平面を評価します。
今回、 pull を3つ定義したうち、実際にサンプリングに使うのは3つ目だけです。それをちゃんと伝えるための coordsel.dat を作ります。
書式はファイル数の数だけ 0 0 1 という行を書いているだけのものです。まあ i 個目の座標をサンプリングに使うなら i 列目を 1 に、使わないなら 0 にするというものです。
ls prod_*_pullx.xvg > pullx-files.dat
ls prod_*.tpr > tpr-files.dat
if [ -f "coordsel.dat" ]; then
rm coordsel.dat
fi
for i in `ls prod_*.tpr`
do
echo "0 0 1" >> coordsel.dat
done
gmx wham -ix pullx-files.dat -is coordsel.dat -tol 1e-2
-tol はもうちょっと頑張ってもいい気はするのですが、デフォルトの基準ではかなり時間をかけないと達成できません。
profile.xvg ができるのでこれを xmgrace で表示します。
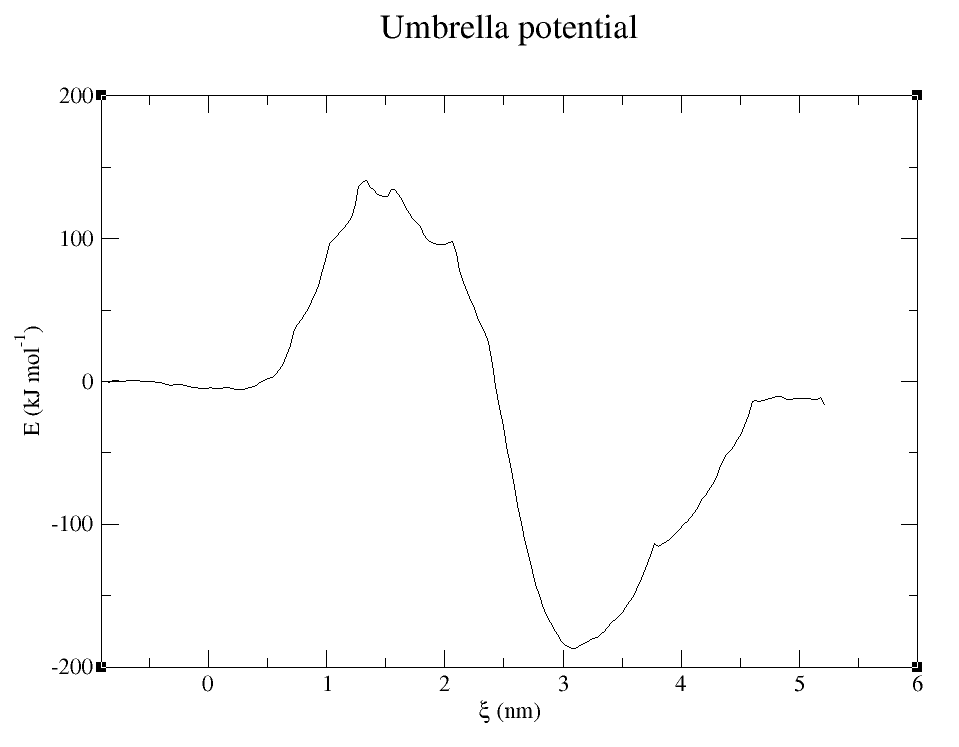
ウチは膜たんぱく質に詳しくないのでこの結果が妥当かどうかはわかりません。
参考までにこの章の初めに使いました、対応するたんぱく質の絵を置いておきます。
データ確認
ちゃんとサンプリングできているかを確認するため、座標 gmx wham では histo.xvg ファイルもできていて、これを可視化すればいいのですが、今回はgnuplotを使ってみます。
XVG形式は最初に @ か # で始まるコメント行で情報が記録されていますので、それをコメント行だとgnuplotに教えてやりつつ、 forループでMD試行ごとのヒストグラムを重ねていきます。
set datafile commentschars "#@"
set xlabel "xi/nm"
set ylabel "Count"
plot for [i = 1:32] "histo.xvg" u 1:i w l lw 2 t ""
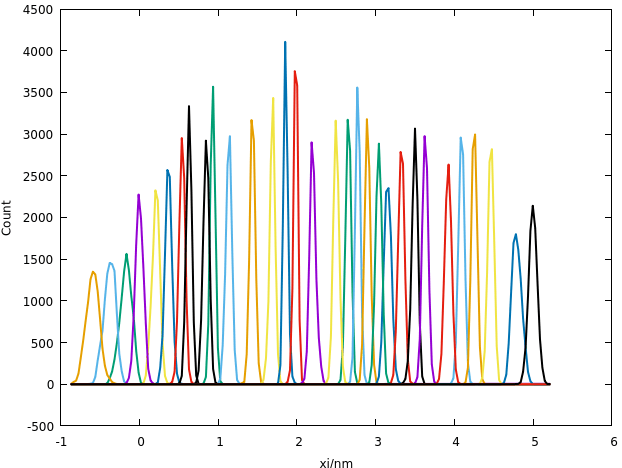
この手法ではヒストグラムを重ねて書いたときに裾が微妙に重なってるくらいがよいと思います。この例ではちょっと断絶してしまっているところがありますね。
参考
なお、実際にカリウムイオンが通過するときはたんぱく質構造が変形することで通過している一方、今回は閉じたままのチャネル中を無理やり通しています。
そのため、障壁がかなり高く評価されています。
Discussion