触媒設計をin silicoで行なって、実際に合成も行なっている論文を読んでみた

0.論文情報
Molecular Field Analysis Using Computational-Screening Data in Asymmetric N-Heterocyclic Carbene-Copper Catalysis toward Data-Driven In Silico Catalyst Optimization
1.論文の概要
本研究では、N-ヘテロサイクリックカルベン(NHC)-銅触媒によるシリルボロン酸のアルデヒドへの不斉カルボニル付加反応に計算機スクリーニングデータを用いた回帰分析を行った。回帰分析にはDFT計算(補足(2))ベースのΔΔG‡の計算値(計30サンプル)をトレーニングデータとして用いて、MFA(補足(3))を実施し、エナンチオ選択性に影響するような構造情報を可視化できるようにした。得られた不斉誘導の機構に関する知見から、分子設計を行ったところ、エナンチオ選択性を向上させる触媒の開発に成功した。
まとめ
実験値を使わず計算で導出されたΔΔG‡(選択性) を用いて分析を行い、効果的な分子設計を行うことができた。
2.問題設定と解決した点(先行研究と比べてどこが凄い?)
先行研究では、機械学習による分子触媒反応の最適化に30個の実験値をトレーニングデータとして分析し触媒設計を行っている(図1A)。しかし正確なデータを常に集められるかというとそうではなく、その値に副反応や実験誤差などの無視できないノイズが含まれている場合がある。そこでDFT計算で得られたデータ、要するに計算値をトレーニングデータとして用いれば、それらのノイズを回避することができる。 そしてデータを収集するための実験が不要であるため、高価で合成が難しい触媒を用いた反応のデータ解析が可能である。
またTS状態の分子場を計算するためにDFT計算を行うことが多いのだが、不斉触媒反応の設計において先行研究が数多くあり信頼できる手法とされている。ある先行研究において、600もの計算値データを用いて回帰分析を行ったが、DFTベースのTSモデリングは計算コストが高く、新たな回帰分析に基づいた手法の開発が求められていた。
本研究では30個の計算値サンプルで触媒設計を行うことができた(図1B)。
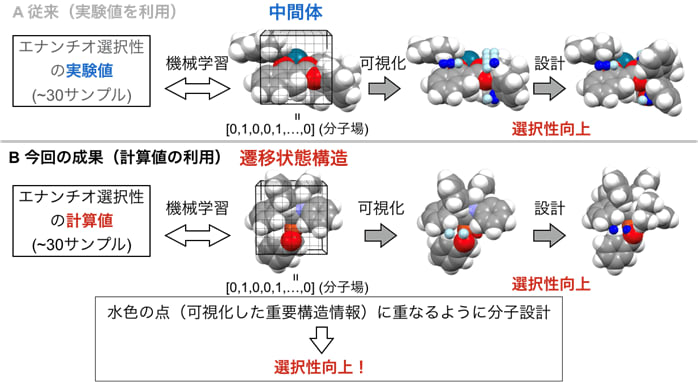
図1 https://www.chem-station.com/blog/2022/02/odx.html より引用
まとめ
- 計算値を用いることでノイズを含まないデータで分析を行い、触媒設計を行うことができた。
- 分析に用いられるDFT計算はデータ数が多いと計算コストも多かったが、データ数を少なく分析することができた。
3.技術や手法のキモ
- この反応ではアルデヒド(Table1参照)が触媒と4中心反応機構を介して進行すると仮定しており、そのTS構造をFigure 2に示す。TS構造計算にDFT計算を用いた。
- データ解析には3D-QSSRアプローチを採用しており本実験ではMFA(Molecular Field Analysis)と呼称している。MFAは計算値とTS構造の分子場との回帰分析を行っている。また分子場としては、立体指標場(補足(4)を採用している。
- 解析を行うとFigure 4のように重要な構造情報がTS構造上に可視化される。青の点は分子構造がそこに位置しているとエナンチオ選択性が高くなる(補足5)ことを示しており、L3S1は分子構造が位置していないL1S1と比べて選択性が高くなっているとわかる。緑矢印付近のフェニレン分子が重なっていたからであり、その理由はiPr基とシリル基(Figure 2参照)の立体反発によって生じると推測している。iPrの代わりにPhを導入することで反発を強くなることを期待してさらに選択性の高い触媒(L4S1)も開発することにも成功した。
Table1 反応と使用する試薬, Figure 2 L3S1の遷移状態構造 S経路とR経路が存在する,Figure 4 TSモデルに可視化された重要構造が書かれていました。
詳しくは、論文をみてください。
まとめ
解析を行い構造上に重要情報を可視化することで、研究者の解釈と合わせてより効果的な分子設計が可能になる。
4.主張の有効性検証
最初に導出された 計算値と実験値は相関を示しており、決定係数(R2値)は0.76となっている。そのほかの構造最適化方法でも同じように重要な構造情報を抽出することができた。
MFAの予測値対計算値のプロットにおいて、初期の18個を用いた場合のΔΔG‡のトレーニングデータはR2:0.78, q2:0.63, 、30個のサンプルだとR2:0.93と, q2: 0.87, Q2:0.79, R2yrandom: 0となっている。(Figure 3参照)
(q2:LOOCV(Leave-one-out Cross Validated)決定係数;Q2:5倍クロスバリデーション決定係数;R2yrandom:R2yrandom:yランダム化により得られた決定係数)
完成した触媒も計算値に対応する実験値は得られている。
L6S1のΔΔG‡:計算値 3.3kcal/mol,実験値 1.7kcal/mol
L7S1のΔΔG‡:計算値 2.1kcal/mol,実験値 2.3kcal/mol
主張の有効性は示されている。
5. 議論すべき点
計算値と実験値を用いた時の違いについての考察を筆者は述べている
(1)計算値と遷移状態構造を用いたMFA
・算出されたΔΔG‡値の信頼性を確認するためには、いくつかの実験が必要だが、 実験なしでトレーニングサンプルを収集することができる
・ΔΔG‡の計算値は、実験値のΔΔG‡と比較して、情報が少ない
・反応機構を把握することが求められる
(2)実験値と遷移状態構造を用いたMFA
・高品質な実験値が必要
・記述子の計算コストが安い
(DFT法による中間構造の最適化の計算コストは、遷移状態計算の計算コストに比べ、 非常に低い)
・実験的なΔΔG‡値には、多くの情報が含まれている
まとめ
計算値があるからといって実験要らずと言うわけではなく、誤差は存在しており計算条件によっても値は変化する。また実験値には実験条件などの情報も多く、解釈性という面でも優れている。どちらの値も特定の強みを持っており、場合によって使い分けることが大事だと考えられる。
6.次に読むべき論文は?
計算値の導出や選択性の予測について詳しく述べられている
MFAの一種のCoMFAについて詳しく述べられている
7.参考文献
著者による日本語のインタビューが載っている
CoMFAについて
著者による総説論文
8.補足(Appendix)
(1)簡単な実験手順
- DFTでΔΔG‡の計算値(3種類の触媒×6基質の18個のデータ)とTS(遷移状態)構造の計算を行う。
- MFAを用いてΔΔG‡の計算値とTS構造の指標場で回帰分析を行う。
- 選択性に影響があると思われる構造情報が可視化されるので、それを元にスクリーニングを行い有効であると思われる構造L4,L5(2種類)を提案した L4,L5を含めた30個(18+2×6)のΔΔG‡の計算値でもう一度同じ作業を行い、L6とL7を提案した
(2) DFT計算
電子密度と個々の電子間に働く引力・斥力相互作用を考慮して安定構造を計算すること。
そこから選択性を計算することができる。
(3)MFA計算
3次元記述子(分子場)を用いて、触媒の活性や選択性の構造的特徴を相関させることができる。そこから不斉触媒構造のどこが立体選択性に重要か可視化もできる。
(4)立体指標場(分子場)の計算
DFT計算により最適化した触媒構造を格子空間に配置する(SIのFigureS2(a)(Ⅲ)参照)。その後任意の原子のファンデルワールス半径を含む単位セルには値1を、それ以外のセルには値0を割り当てた。(図5参照)

図5 立体指標場の計算
Discussion