【Kaggle】【銀賞】DNA-Encoded Library(DEL)を使ったAI創薬
本記事は創薬コンペにチームで出場し、銀賞を取れたので、その体験をまとめています。
以下に英語版があるので、そちらを主に書き換えています。
27th Place Solution (8th in Public): 1DCNN for share and ChemBERTa for non-share
対象読者
- 機械学習の基礎はわかり、AI創薬に興味があるエンジニア
- 普段はWetを行なっているが、dry研究に興味がある研究者
- AI創薬の手法を知りたい方
- Kaggleに挑戦したい人
など
コンペ
NeurIPS 2024 - Predict New Medicines with BELKA
背景
DNA-Encoded Library(DEL)は化合物スクリーニング手法の一つであり、大規模化合物ライブラリから標的タンパク質に結合する化合物を取得できることから、創薬分野で注目されています。
各化合物に対応するDNAが低分子化合物に結合しているため、DNAを読むことで、化合物の同定が可能です。今回のコンペでは以下の図のように真ん中にトリアジンコアがあり、それに対して三つの異なるbuilding blockを持つ化合物を対象としていました。
trainデータには三つのbuilding block、化合物全体の構造を示すSMILEとBRD4, HSA, sEHというタンパク質に結合するかどうかのラベルが与えられ、testデータの化合物の結合を予測するものでした。この予測により、実際に実験をしなくても標的の化合物に結合する化合物がわかるので、コスト削減につながることが期待できます。
目的
DELを使って得られたデータを学習して作った機械学習モデルを使って、BRD4, HSA, sEHという三つのタンパク質に対する化合物の結合確率を予測します。
Challenge
Challenge 1:データ量が非常に多かった。1つのタンパク質につき、9800万 training 36万 test dataを使用。
Challenge 2:trainデータにはトリアジンコアであるものだけだったが、テストデータにはトリアジンコアでないコアのものが5割くらい入っていた。
我々の解決策
Challenge 1に関しては、サンプリング数を少なくして対応しました。また1DCNNに関してはSMILESを数値に変換も行いました。
Challenge 2に関して、まずtestデータのトリアジンコアのもの(=share)には1DCNN、トリアジンコアでないもの(=non-share)にはChemBERTaを使い、それぞれをチューニングしてい来ました。
トリアジンコアのもの(=share)
モデル:1DCNN
1DCNNは以下のアーキテクチャーです。
こちら の記事を参考にしてください。
- 入力層:インプットデータを受け取る。
- 畳み込み層:1D畳み込みフィルターを適用して特徴を抽出。
- 活性化層:ReLU関数を使用して非線形変換を行う。
- プーリング層:最大プーリングでデータの次元を削減。
- 全結合層:抽出された特徴を用いて最終的な予測を行う。
なおデータ量が多い問題に関しては、文字から数値への変換により、計算を楽にしていた。

ディスカッションで公開されたコードについて、下記の項目を改変しました。
全体図は以下の感じ
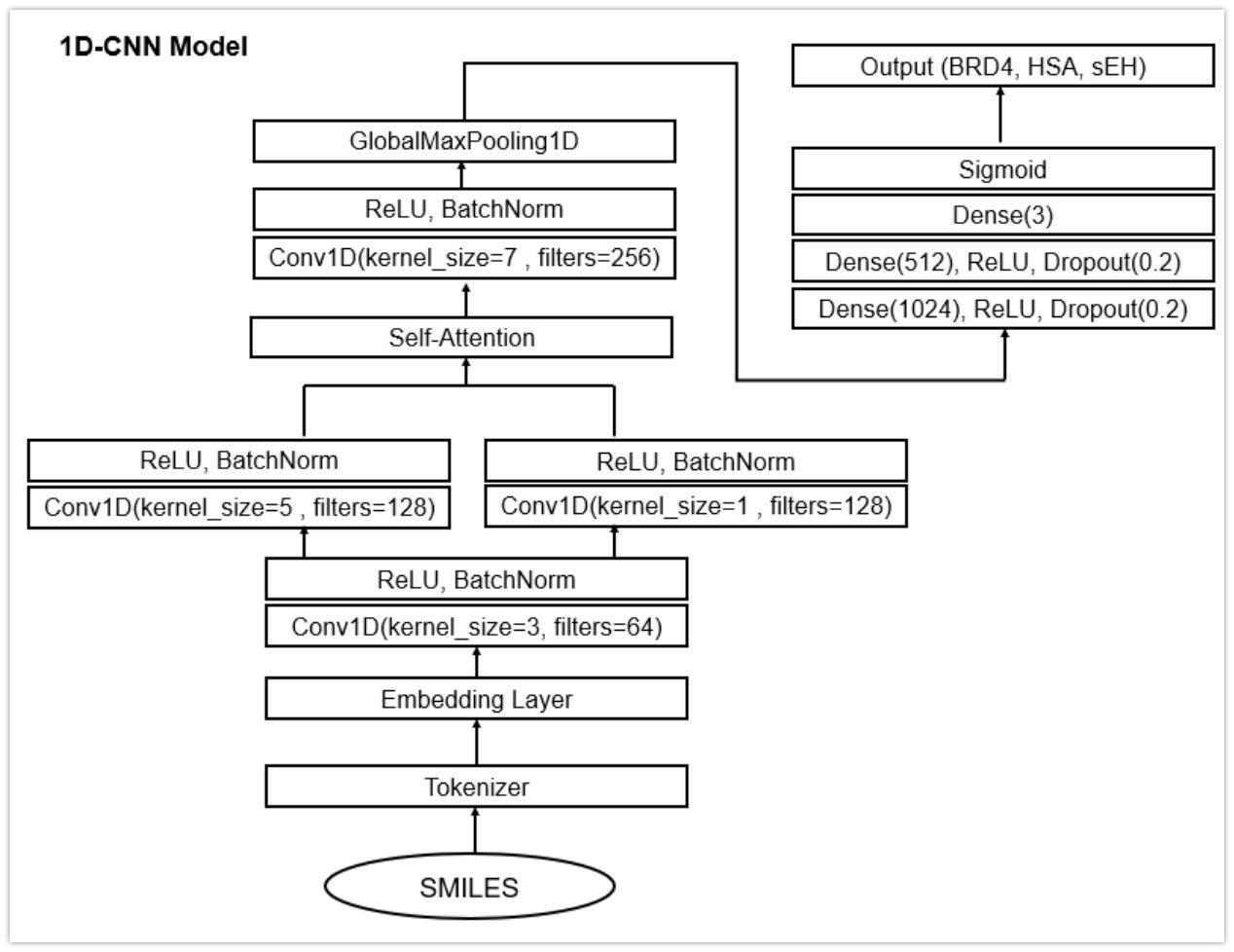
なおデータ量が多い問題に関しては、文字から数値への変換により、計算を楽にするという工夫がなされていた。
モデルの工夫
(画像は変更前の公開ノートブックの情報です。)
-
フィルターの増加:
各層でフィルター数を32から64に倍増させて、より多くの特徴を抽出。
(以下のNUM_FILTERSを64に)

-
強化された畳み込み層:
'same'パディングと様々なカーネルサイズを使用して、出力サイズを維持しつつ詳細な特徴を捉える。
(以下のpaddingをsameに)

- バッチ正規化:
各層の出力を正規化して学習の安定化と加速化を図る。
以下の矢印にx = tf.keras.layers.BatchNormalization()(x)を入れる。

-
introduced shortcut connections and Attention mechanism
深いネットワークでも効果的に学習するために導入。
第二層の箇所に以下のshortcut connectionコードを追加
x2_shortcut = tf.keras.layers.Conv1D(filters=NUM_FILTERS*2, kernel_size=1, padding='same', activation='relu')(x1) x2 = tf.keras.layers.Add()([x2, x2_shortcut])また残差接続の後にの部分にAttenstionレイヤーも追加
x = tf.keras.layers.Attention()([x, x])xをクエリ、キー、バリューとして同じ入力を用いてAttenstionを適用しています。これによりデータ内の重要な特徴に焦点を当てることができます。
これらの変更で勾配消失問題を解決できます。
勾配消失問題とはニューラルネットワークが深くなるほど勾配が小さくなり、重みの更新が十分に行われない結果、学習が停滞するという現象のことです。

-
ドロップアウト率の調整と全結合層の変更
0.1から0.2に設定して過学習を防いでいます。。
→各訓練ステップで、設定された割合(この場合は0.2)のノードをランダムに無効化し、モデルが特定のパスに依存するのを防ぎます。これにより、モデルの汎化性能が向上します。全結合層の変更
全結合層の数を増やすと、モデルの表現力が向上し、複雑なパターンを学習できるようになります。また、relu関数を噛ませているので、非線形性が増加することで、より高度な関数を学習可能です。一方で過学習のリスクもあるため、ドロップアウトや層を減らすなどをしています。以下の部分の0.1を0.2にしています。さらに全結合層を2層にしている。

訓練の工夫
-
エポック数の増加
20から30に増加させて学習期間を延ばしています。
エポック数が多いほど、モデルはデータセットを繰り返し学習し、パターンをより深く理解します。

-
ReduceLROnPlateauのpatienceを減少
ReduceLROnPlateauは、検証損失(val_loss)が改善しない場合に学習率を減少させるためのコールバックです。
検証損失(Validation Loss)は、モデルが検証データセット(トレーニングに使用しないデータ)に対してどれだけ誤差があるかを示す指標です。これにより、学習プロセスが停滞した際に適応的に学習率を調整し、モデルの最適化を促進します。
5から3に短縮して早期に学習率を調整しています。

- 軽度の過学習を許容:
トレーニングデータの特徴を深く学習し、予測性能を向上させています。
トリアジンコアでないもの(=nonshare)
モデル:ChemBERTa-10M-MTR ベースはこちら
ChemBERTaに対してもっともnonshareに対する予測が良かったため、利用しました。これは事前学習による影響が多いと考えています。
ファインチューニング(事前学習したモデルを今回の目的のために変更すること)
・1タンパク質に対して、400万のサンプルを使って、モデルを学習しました。(全てのタンパク質ではメモリが不足したため)。
・1タンパク質に対して、それぞれモデルを作成しました。(3つのタンパク質の予測をするのに一つのモデルではない)
・sEHに関しては外部データも混ぜて使いました。
・過学習を避けるため、ChemBERTaの最初の12層のうち3層の重みを更新しませんでした。(全部で9層で学習)
Cross-Validation (交差検証)strategy
交差検証は、モデルの性能を評価するためにデータセットを複数の部分に分割し、各部分を使ってモデルを複数回訓練および評価する手法です。この方法は、モデルの一般化性能をより正確に評価するために使用されます。
Shareに関しては、StratifiedKFoldを用いました。
non-shareに関しては、以下のノートブックを元にバランスが保たれるように作りました。
他うまくいかなかったが、頑張ったもの
スタッキング
スタッキングは、複数の機械学習モデルの予測結果を組み合わせて、より強力な予測モデルを構築する手法です。これにより、各モデルの強みを活かし、全体の性能を向上させることができます。 上記のモデルの前に様々なものを作成したので、それらのデータをスタッキングさせました。
モデル
共有データ(Share):1DCNN、LGBM2(ECFP6を使用して10Mデータで学習)、ChemBERTa
非共有データ(Non-Share):ChemBERTa、1DCNN、LGBM1(morgan4+rdkit+descriptorsを使用して全データで学習)、LGBM2
Stackingのやり方
- 各モデルを使用して、クロスバリデーションの各フォールドで予測を実行し、それを x としました。
- コンペティション主催者が提供したトレーニングデータのbind値を y としました。
- カテゴリデータに強いCatboostをスタッキングモデルとして使用しました(実際には bb を入力として使用しませんでしたが)。
- 提出用のバインド値を予測するために、各モデルによって予測されたテストデータのバインド値を x として使用しました。
- 最後に、共有データと非共有データの予測をマージしました。
結果
ShareとNonshareに対して、それぞれstackingしたものを組み合わせましたが、スコアの向上はありませんでした。
Share コード
Nonshare コード
ほか細々な検討
- Scaffoldを除いて、building blockのみを一つにまとめてChemBERTaで予測しました。。
→スコアの上昇は見られませんでした。building blockのみを別々に分けて学習したらまた違っていたかも。
コード - 化学特性(分子量、LogP、分子内の水素原子数、水素結合受容体、回転可能な結合の数、分子の極性表面積)を特徴量として追加しました。
→重要度が低い特徴量(分子内の水素原子数、水素結合受容体)を削除すると、少し改善されました。(LBスコアは0.452 => 0.486)
- HSAに関してはlogPが結構使えるらしい。
- フィンガープリントを比較。ECFP4, ECFP6, avalon, maccs, rdkit, ecfp4+maccs, ecfp6+maccs, rdkit+maccs
→一番ECFP6が良かったです。
※RDKitにはオープンソフトウェアとしてのRDKitとフィンガープリントのRDKitフィンガープリントがあるので、文脈によって判断する。

(@kyu999_mochiさん作成)
- 分子フィンガープリントのMalformer埋め込みに置き換え
分子フィンガープリントを使っていた部分を、より高度なMalformer埋め込みに置き換えています。
Malformer埋め込みは深層学習を用いて分子の複雑な特徴を捉え、より良いパフォーマンスを提供する可能性があるためです
- SMILES enumeratorを用いて生成された拡張データの効果を評価しました。
→あまりうまくいかきませんでした。
- ChemBERTa(10M SMILES)と更新モデルChemBERTa2(77M SMILES)の比較
→ChemBERTa(10M SMILES)の方が良かった。
- Autodock vina(PyRx)を使用して結合力予測→新しい特徴量としたい
→すでにbindの有無と結合力の関係があまり出ませんでした。
Autodockのpython wrapperがあるらしいです。コード上で結合力を予測できるので、便利そうです。
コンペを出た感想
私は元々mRNA displayを使ったペプチドのスクリーニングを研究していたので、その低分子バージョンのDELの機械学習を学べたことはとても良い経験でした。ペプチドでも良い教師データがあれば、参考にしてモデルを作ってみたいと考えています。特にHSAに関しては血中で最も豊富なタンパク質であり、体内で候補薬を吸収し、それらを標的組織から隔離する役割を果たすことが多いため、応用性が高いと思っています。
今回のコンペでは、タンパク質の立体情報を特徴量に入れるということはどのチームもやっていなかったように思ええます。また分子ドッキング手法の値を特徴量に入れることも試みたが、うまくはいきませんでした。今後このような情報も取り入れることができるようになれば、もっと結合力予測がやりやすくなる可能性が高いと思いました。
また今回のコンペではDEL化合物ライブラリはすでに決まっており、それに対する学習と未知の化合物の予測だった。当たり前かもしれないが、scaffold(トリアジンコア)を変えるとbindingの仕方がもちろん変わることが多いと思います。それは機械学習でも同じということを感じ取れました。今回は分子の構造上、フレキシビリティの高い分子を扱っていたので、これがよりrigidな構造での化合物ライブラリのデータであればもっと簡単にモデルを構築できたかもしれないということを感じました。wet研究者とdry研究者との協力するにあたり、この点は気をつけないといけないと感じました。
今回のコンペでは様々なモデル、改変の仕方を勉強させていただきました。私のような素人には一見どのパラメータをいじれば良いかということは検討がつかなかったですが、@shina_kaggleさんのやってくださった1DCNNの改変の仕方は大変勉強になりました。またもっともsubmissionの回数が多く、そのパワフルさにとても感銘を受けました。
また全体の方針の決め方に関して、機械学習の総合的な知見を有する@ne_gi_chi__さんの意見がとても勉強になりました。モデルを一から自分で作るなど、そのコーディング力の強さにはとても驚きました。ChatGPTなどの生成AIでコードはある程度かけるようになりましたが、一から書く必要性をとても感じることができました。また外部GPUを使えるということで、今回のコンペはデータ勝負みたいなところもあり、PCのスペックの重要性も実体験できました。
様々な外部データや論文の情報をいち早く取り入れ、検討してくださった@kyu999_mochiさんの情報収集力とそれを実現する能力にとても感銘を受けました。私自身もまだまだ論文を読んで試すみたいな経験は少ないので、今後頑張っていきたいと思っています。またうまくいかなかったもののフィンガープリントの比較やデータ拡張のアイデアは個人的に面白かったです。Autodockのpython wrapperについては今後使っていきたいと思っています。
また今回は私@shizuku50312776がXなどで呼びかけてチームを結成させていただきました。コンペが始まった直後からチームを組んだので、様々なアプローチを試すことができ、モデルごとに作業分担、ディスカッションをあまりこぼすことなくウォッチできたのが良かったです。3ヶ月コンペ漬け+初心者だったので、結構大変でしたが、それに見合うだけのリターンはありました。エンジニア歴の浅い方でも思い切って、チームを組むことをお勧めします。
最後になりますが、一緒に出てくださった@ne_gi_chi__さん、@shina_kaggleさん、@kyu999_mochiさんに心より感謝申し上げます。私自身わからないことが多かったですが、その都度丁寧に返答してくださり、とても勉強になりました。チームメンバーに恵まれたので、とても良かったです。ありがとうございました!
Discussion