GROMACSのPBC周りのもろもろ

はじめに
MD計算では多くの場合、周期境界条件(PBC)を適用させます。つまり、原子がシミュレーションボックスの端に到達すると、もう片方の端から出てくるようになっています。
計算上は、これで問題はないはずです。しかし、解析時やVMDなどによる視覚化の際には分子がPBCを超える際にワープしたり、途切れたりすることが面倒な問題の原因になります。
GROMACSでのこの辺の挙動がよくわからず、またググっても微妙に欲しい情報がすぐに出てこなかったので、備忘録を兼ねてまとめてみました。
検証バージョン
gromacs2022.2
ほかのバージョンでもおおむね変わらないとは思います
内容
水溶液中のグルコース分子のMDデータを例に説明していきます。
なお、データの生成方法はこちらになります。
エネルギー最小化、nvt平衡化ののちに、本計算100 ns(5ns x 20)を行っています。
以降の説明では本計算部分のトラジェクトリのみを用います。
トラジェクトリ生成コード
#!/bin/bash
# 元コードを適当に簡素化しているので少し間違っている可能性があります
set -e
Lig="Glc"
SysDir="testdir/${Lig}"
MdpDir="testdir/mdps"
InputPdb="${SysDir}/Glc_final.pdb"
MinMdp="${SysDir}/min.mdp"
MinGro="${SysDir}/min.gro"
MinPrf="${SysDir}/min"
Top="${SysDir}/top.top"
Ind="${SysDir}/index.ndx"
SimLength=100
EachLength=5
MaxPrdStep=$(( $SimLength / $EachLength ))
# minimization
gmx_mpi grompp \
-f $MinMdp \
-c $InputPdb \
-p $TopFile \
-o min.tpr \
-maxwarn 3
gmx_mpi mdrun \
-s min.tpr \
-o min.trr \
-c min.gro \
-e min.edr \
-g min.log
# nvt
gmx_mpi grompp \
-f ${MdpDir}/nvt.mdp \
-c ${MinGro} \
-p $Top \
-o nvt.tpr \
-n $Ind \
-maxwarn 3
gmx_mpi mdrun \
-s nvt.tpr \
-o nvt.trr \
-c nvt.gro \
-e nvt.edr \
-ntomp 12 \
-g nvt.log \
-v
# run production md
input_gro=nvt.gro
for i in {001..020}; do
prdname=prd${i}
gmx_mpi grompp -f ${MdpDir}/npt.mdp -o ${prdname}.tpr -c ${input_gro} -p ${Top} -n ${Ind} -maxwarn 3
gmx_mpi mdrun -v -deffnm ${prdname}
input_gro=${prdname}.gro
done
イメージ画像(最小化前)
このデータ・コマンドで生成されたトラジェクトリデータを使っていきます。
1. mdrunコマンドで出力される.xtc, .groのPBC
まず、mdrunで生成されたprd001.xtcなどのデータにおいてPBCの取り扱いがどうなっているのか見てみます。今回、5 ns x 20ファイルに分割されているので、一度trjcatコマンドで20ファイルをすべてくっつけます。
# count the number of xtc files to combine
no_xtc=`ls -1 prd0[0-9][0-9].xtc | wc -l`
# concatenate xtc files
for i in `seq 1 $no_xtc`;do
echo c
done | gmx_mpi trjcat -f prd0[0-9][0-9].xtc -o prd.xtc -settime
くっつけたトラジェクトリファイルprd.xtcの中身をcheckコマンドで確認します。
Command line:
gmx_mpi check -f prd.xtc
Checking file prd.xtc
Reading frame 0 time 0.000
# Atoms 3632
Precision 0.001 (nm)
Last frame 1000 time 100000.000
Item #frames Timestep (ps)
Step 1001 100
Time 1001 100
Lambda 0
Coords 1001 100
Velocities 0
Forces 0
Box 1001 100
3632原子、1001フレームあるのがわかります。
今回は0.1 ns刻みにしているので、最初の構造(=nvt平衡化の最終構造)を含め、想定通り100 ns分ちょうどあることがわかりますね。
本筋とは関係ないですが、trjcatの仕様について少し補足しておきます。
補足
個別の.xtcファイルの中身はこんな感じです。
Command line:
gmx_mpi check -f prd001.xtc
Checking file prd001.xtc
Reading frame 0 time 0.000
# Atoms 3632
Precision 0.001 (nm)
Last frame 50 time 5000.000
Item #frames Timestep (ps)
Step 51 100
Time 51 100
Lambda 0
Coords 51 100
Velocities 0
Forces 0
Box 51 100
原子数は一緒ですが、フレーム数は51フレームで、5 ns相当あります。ここで、51 x 20ファイル = 1020フレームとなるので、すべてくっつけたprd.xtcのフレーム数とつじつまが合いません。
実はtrjcatコマンドでc連打すると、2ファイル目以降では、最初のフレームが無視され、前のファイルの最終フレームが採用されます。前のステップの.xtcの最終構造は次のステップの.xtcの初期構造と全く同じなので、この仕様は自然なものではあります。trjcatを使いたい場面によってはこの仕様に意外と詰まったのでここに書き残しておきます。
では、このトラジェクトリprd.xtcをVMDで可視化してみます。トポロジーには初期構造であるnvt.groを使います。
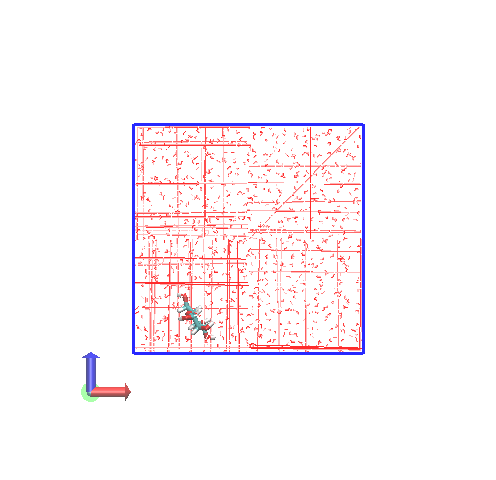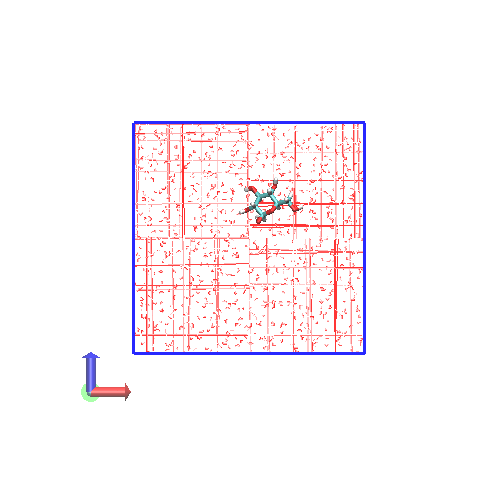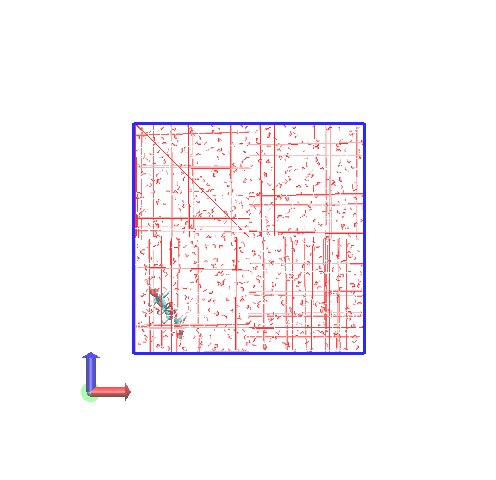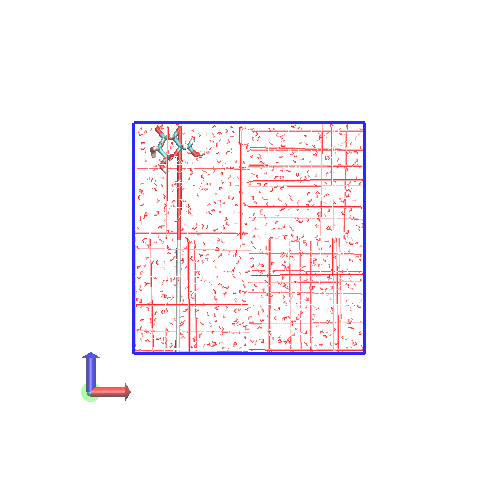
赤線(水分子)やLicoriceで表示されるグルコース分子が途切れまくっているのがわかります。
また、青線で示されるシミュレーションボックスの外に出ている原子が全くないのがわかります。
このように、gmx mdrunで生成されるトラジェクトリ.xtcでは、すべての原子がボックスの中にあるように平行移動されています。 結果として分子が途切れているように表示されてしまいます。
また、gmx mdrunを実行終わる際、最後のフレームのデータが.groファイルとして出力されます。このファイルについても、周期境界がどうなっているのか見てみます。
今回、20個の.groが出力されているので、すべてをVMDで一気に読み込んで、トラジェクトリのようにして表示します。
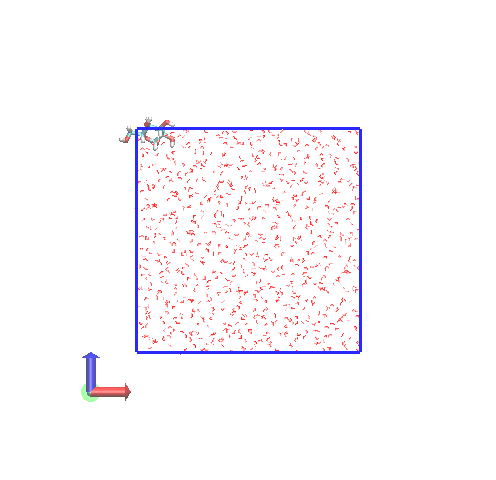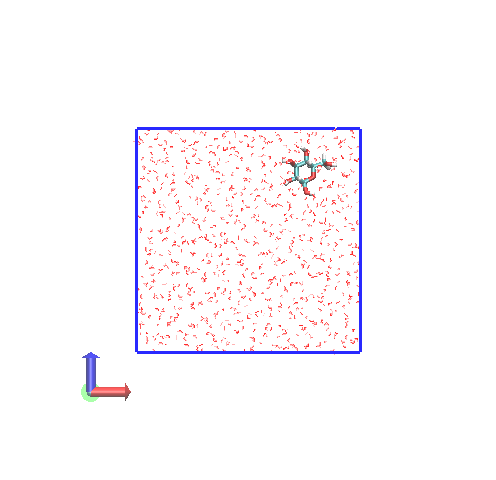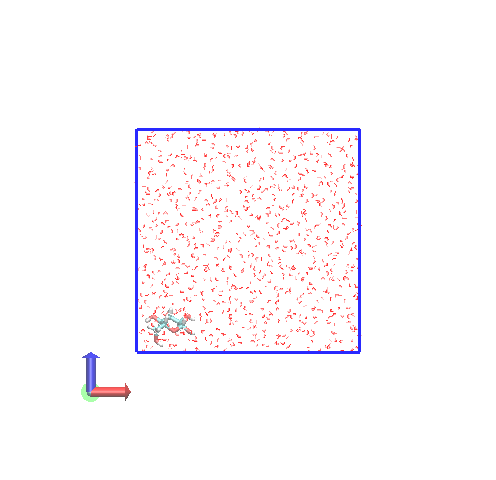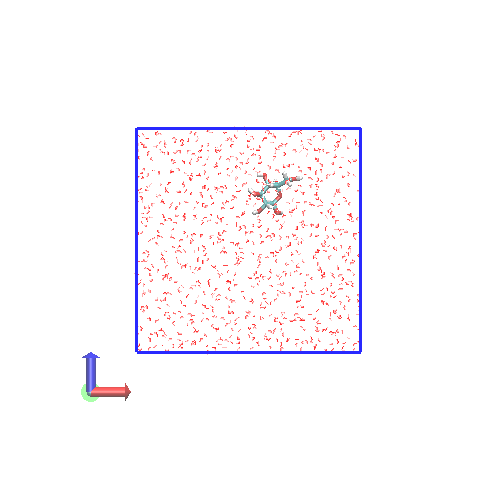
今度は、ボックスを横切るような赤線が見えていません。グルコース分子も途切れていません。つまり、 計算終了時に出力される.groファイルでは、ボックスを横切って途切れている分子が修正されています。 この修正は、.topの中にある[molecules]単位で途切れがないように原子を平行移動させていると思われます。この場合は、原子によっては、ボックスの外に出ているものもあるということになります。
2. trjconvによるPBCの処理
前節でみたように、生の.xtcファイルでは分子の途切れやジャンプが見られます。
これらの修正には、gromacsのコマンドgmx trjconvを利用することができます。
このコマンドにはたくさんのオプションがありますが、PBCに関係するのは-pbcです。
このオプションへの引数として与えられる値について、ドキュメントに記載があります。
molputs the center of mass of molecules in the box, and requires a run input file to be supplied with -s.resputs the center of mass of residues in the box.atomputs all the atoms in the box.nojumpchecks if atoms jump across the box and then puts them back. This has the effect that all molecules will remain whole (provided they were whole in the initial conformation). Note that this ensures a continuous trajectory but molecules may diffuse out of the box. The starting configuration for this procedure is taken from the structure file, if one is supplied, otherwise it is the first frame.clusterclusters all the atoms in the selected index such that they are all closest to the center of mass of the cluster, which is iteratively updated. Note that this will only give meaningful results if you in fact have a cluster. Luckily that can be checked afterwards using a trajectory viewer. Note also that if your molecules are broken this will not work either.wholeonly makes broken molecules whole.
要約すると、mol, res, atomはそれぞれ分子、残基、原子レベルで計算した重心が箱の中に入るように平行移動するオプションで、nojumpは初期構造から各原子が周期境界を横切る際にジャンプして反対側に行くことがないようにするもの、clusterはiterativeに原子座標を動かしていってcluster内に入っている原子たちが重心に最も近くなるようにするもので、wholeは、単にpbcで途切れた分子を連続的になるように修正するものということです。
また、関連するオプションとして-urがあり、次のように説明されています。
Option
-ursets the unit cell representation for optionsmol,resandatomof-pbc. All three options give different results for triclinic boxes and identical results for rectangular boxes.rectis the ordinary brick shape.tricis the triclinic unit cell.compactputs all atoms at the closest distance from the center of the box. This can be useful for visualizing e.g. truncated octahedra or rhombic dodecahedra. The center for optionstricandcompactistric(see below), unless the option-boxcenteris set differently.
引数としてはrect, tric, compactがあり、これらの指定は-pbcにmol, res, "atom"hを指定した場合のみ有効であるということです。基本的に直方体以外の形のボックスの場合に使うオプションに見えます。一般的な直方体ボックスだとrectでもcompactでも変わらなそうな気がします。
なお、デフォルトでは-ur rectです。
今回は、prd.xtcに対して各オプションをかけてトラジェクトリデータを操作してみることで、実際の操作内容を確認してみます。
コマンド
#!/bin/bash
# コードを簡単にするため、無効なオプションセットも含めて実行
for pbc_option in mol res atom cluster nojump whole; do
for ur_option in rect tric compact;do
suffix="${pbc_option}_${ur_option}"
echo "Processing ${suffix}..."
if [[ "$pbc_option" == "cluster" ]];then
Input="GLC System"
else
Input="System"
fi
echo ${Input} | gmx_mpi trjconv -f prd.xtc \
-s prd001.tpr \
-o prd_${suffix}.xtc \
-pbc ${pbc_option} \
-ur ${ur_option} \
-n index.ndx
done
done
まず、出力内容がどれだけ違うか見てみます。
md5sum prd_*
結果はこんな感じになります。
6524d185fdfe63ade3ed09a38f9cd237 prd_atom_compact.xtc
36a6e8572dfe6a077e819bda084dc318 prd_atom_rect.xtc
36a6e8572dfe6a077e819bda084dc318 prd_atom_tric.xtc
ecf1b97b3fd5afe728e858bca0b6d629 prd_cluster_compact.xtc
ecf1b97b3fd5afe728e858bca0b6d629 prd_cluster_rect.xtc
ecf1b97b3fd5afe728e858bca0b6d629 prd_cluster_tric.xtc
60ffab865af2bbf608c9b7bd46b8b7a0 prd_mol_compact.xtc
ac1b226c78ef1a9f90f5adf8c281f512 prd_mol_rect.xtc
ac1b226c78ef1a9f90f5adf8c281f512 prd_mol_tric.xtc
b84693d50e0af98344584e758329fc2c prd_nojump_compact.xtc
b84693d50e0af98344584e758329fc2c prd_nojump_rect.xtc
b84693d50e0af98344584e758329fc2c prd_nojump_tric.xtc
2e5f7b052e4064734dfa80e7f8d89d5d prd_res_compact.xtc
e5027dfbd6047e55b39bac162eb0a641 prd_res_rect.xtc
e5027dfbd6047e55b39bac162eb0a641 prd_res_tric.xtc
b435e85ee8a264f153b65964c8720c59 prd_whole_compact.xtc
b435e85ee8a264f153b65964c8720c59 prd_whole_rect.xtc
b435e85ee8a264f153b65964c8720c59 prd_whole_tric.xtc
確かに-pbc whole, -pbc nojump, -pbc clusterでは-urオプションの中身に関わらず出力された.xtcの内容が変わらず、ドキュメント通りになっています。
こんな感じのwarningも出ました。
WARNING: Option for unitcell representation (-ur tric)
only has effect in combination with -pbc mol, res or atom.
Ingoring unitcell representation.
また、-ur rectと-ur tricでは結果が同じになっています。今回は、今回は直方体のボックスなので-ur tricの場合はオプションが無効化されているものと思われます(warningも出てたはずですが、見逃してしまいました...)。
では、各トラジェクトリをVMDで可視化してみます。
組み合わせが多いので、-pbcオプションごとにまとめて順番に見ていきましょう。
各画像左から順に-ur compact, -ur rectです。
tricはrectと一緒なので省略します。
なお、左右に並べてみたものの、-ur compactと-ur rectの違いはよく分かりませんでした...
ハッシュ値が違うのでどこかしら違いはあるはずなのですが...
まず、-pbc mol
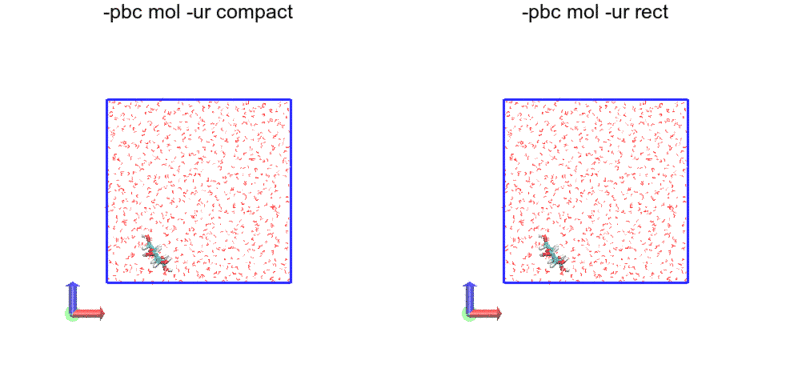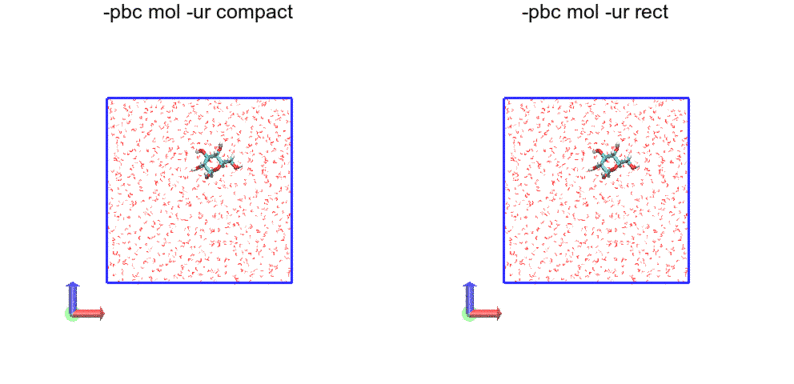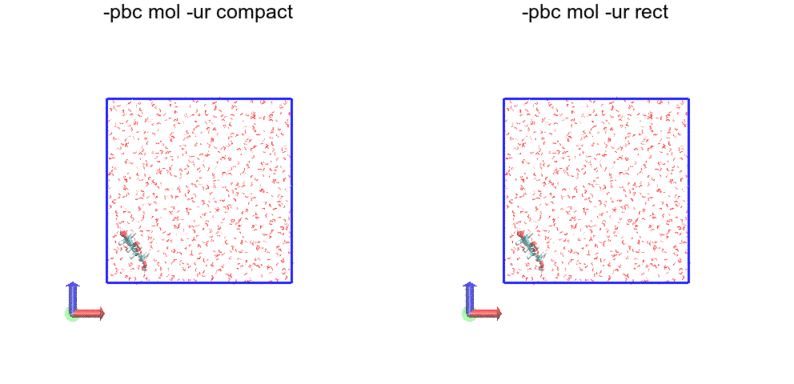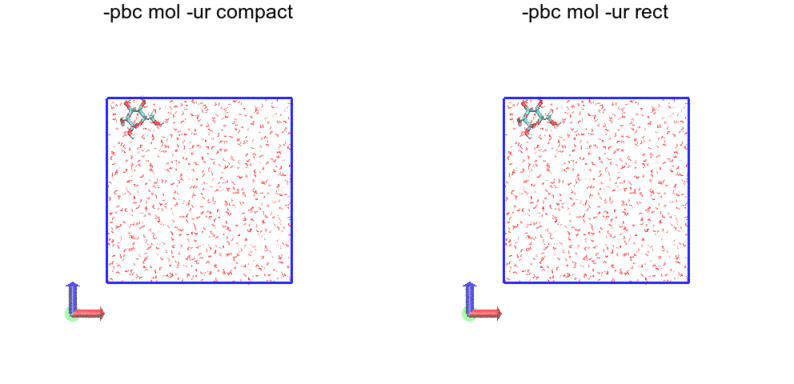
分子が途切れていないのがわかります。解析内容によってはこれで十分ですね。しかし、ボックスの境界に達するとジャンプするので、座標データが不連続になってしまいます。
次に、-pbc res
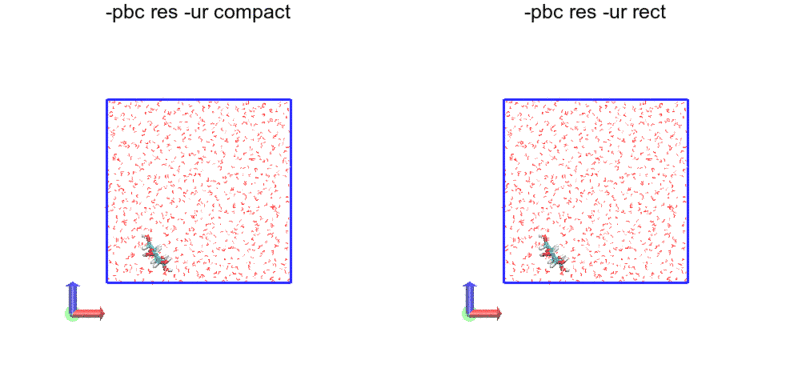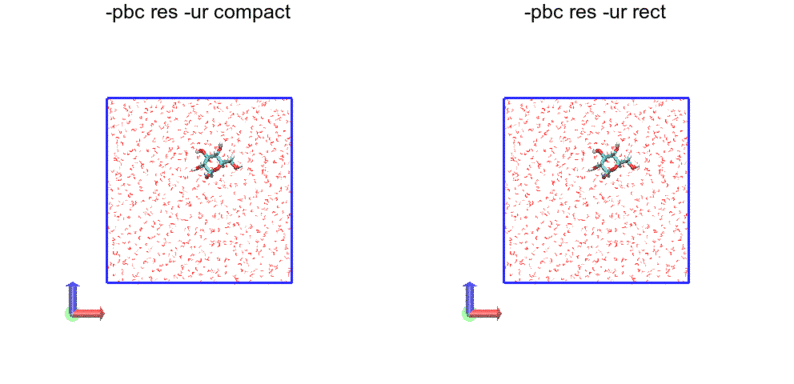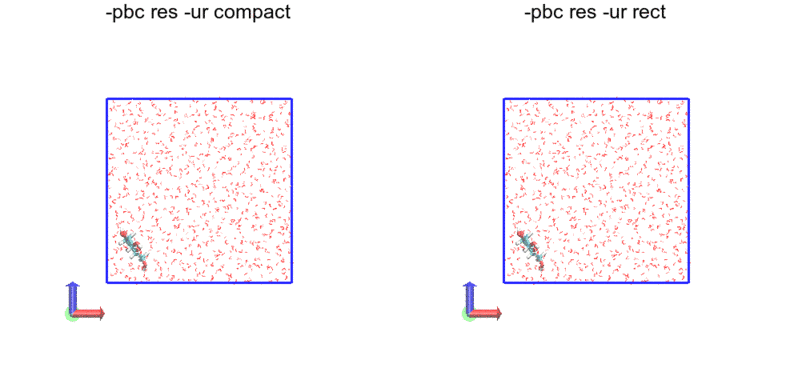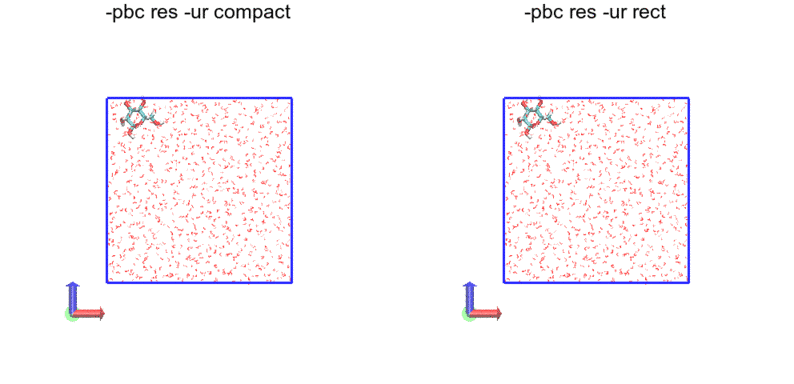
今度は、残基単位(resnameとresidで判定している?)で途切れがないようになっています。今回利用した力場では、グルコースは二つの残基からなっているため、途中グルコース分子内で途切れている箇所があります。一方、水分子は一分子が一つの残基からなるため、途切れていません。
続いて、-pbc atom
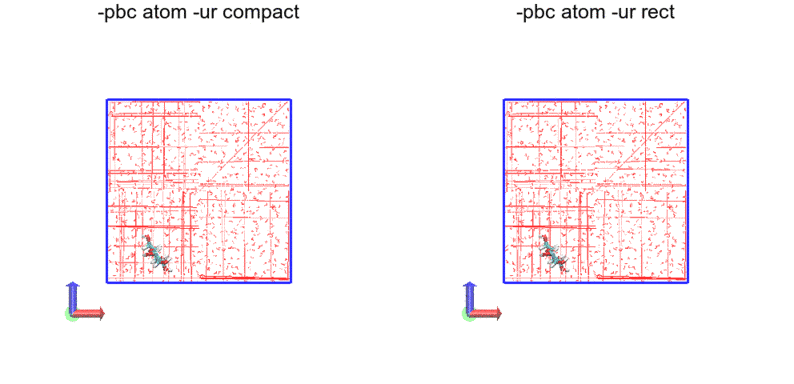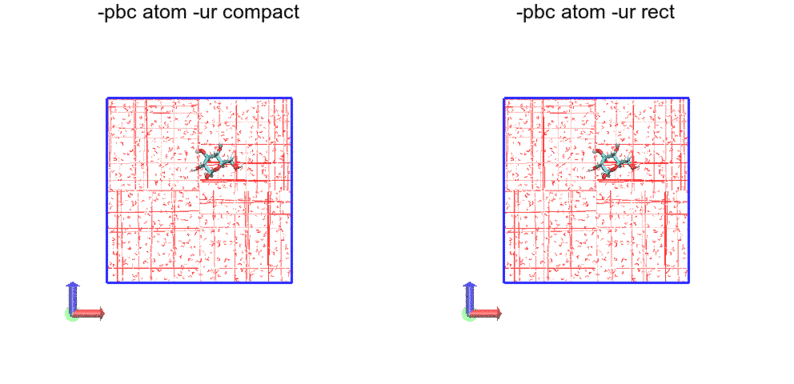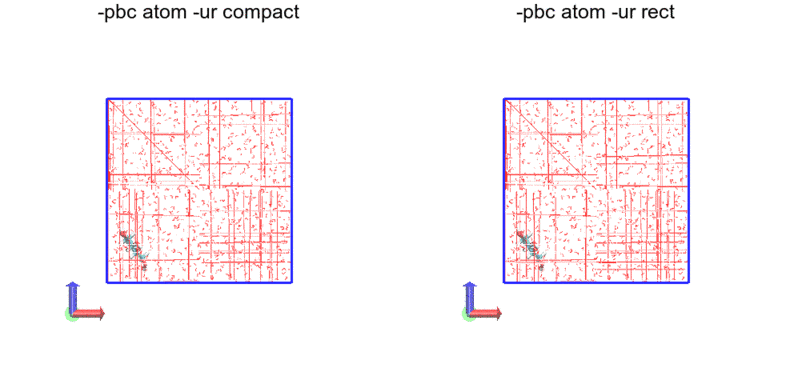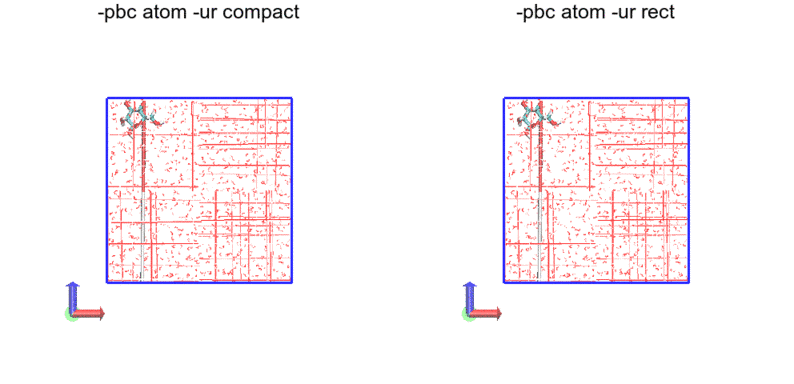
今度は原子単位です。つまり、全原子が箱の中に納まるようにしているだけです。したがって途切れもジャンプもあります。
-pbc cluster

一見、molと似ていますが、trjconv時にgroup for clusteringで指定したグルコース([GLC])のみが修正されており、そのほかの水分子は修正されておらず、もとのトラジェクトリ同様途切れまくっています。
アルゴリズム的にはiterativeにグループ内の原子座標をアップデートするようです。いまいちよくわからないので、mddtrajで修正前の座標データと比較してみます。
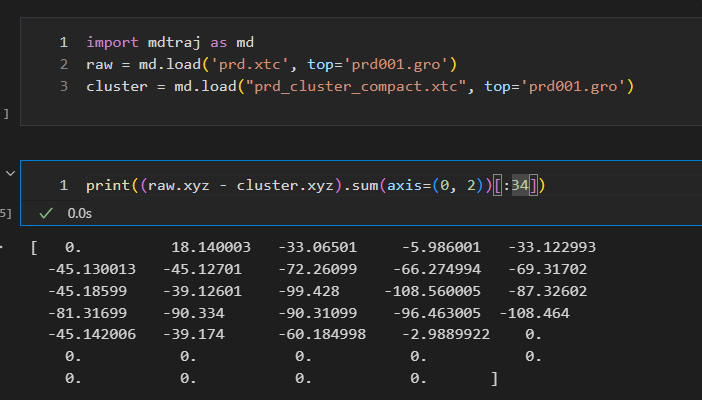 グルコースは24原子からなりますので、25個め以降が0ということは、実際に水分子部分の座標は変更がないことがわかります。また、グルコース部分も、最初の原子には変更がありません。
グルコースは24原子からなりますので、25個め以降が0ということは、実際に水分子部分の座標は変更がないことがわかります。また、グルコース部分も、最初の原子には変更がありません。
つまり、操作の中身としては「選択したグループの最初の原子を基準に、グループ内の他の原子を少しずつ動かしてグループ内で途切れ(=ボックスサイズの半分以上離れている)のないように修正する」ということでしょうか?でもこの操作って1個ずつ最初の原子に近いところに動かしていくだけになるので、iterativeという言葉のイメージと会わないような気がします。もしかすると、今回たまたま最初の原子が固定されてただけで、もう少し複雑なやり方で処理しているのかもしれません。
このオプションでやりたいこと的には、正直-pbc molで十分な気もしますが、他の分子を修正しない分、多少処理時間が短いなどあるのでしょうか...?
-pbc whole

分子が途切れのないように修正されるという点で、-pbc molと違いがないように見えます。ドキュメントを読んだ限りでは、「各分子の重心がボックス内に来るようにする」という操作をするのがmol, しないのがwholeであるように思われます。では、この操作をしない場合、分子ごとに、どの原子を基準に途切れがないよう修正するのでしょうか?(ボックスの両端に分子がまたがっている場合、どちらに合わせるかは自明でないように思います)
再度mdtrajを使って、clusterと比較してみます。
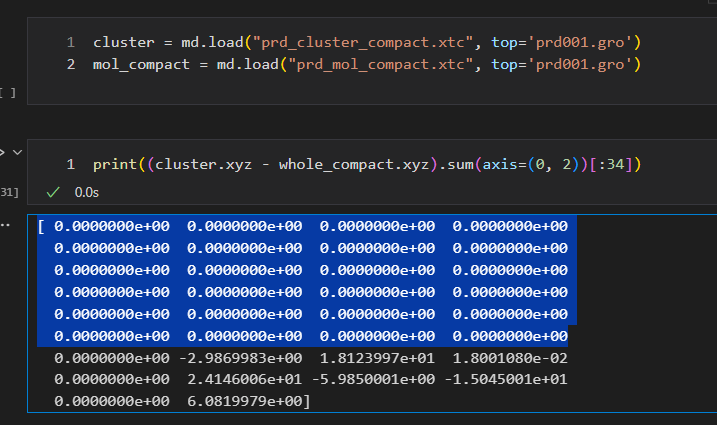
グルコース部分(最初の24原子)の座標が全く一緒ですね。つまり、最初の原子を基準にしているということのようです。
-pbc nojump
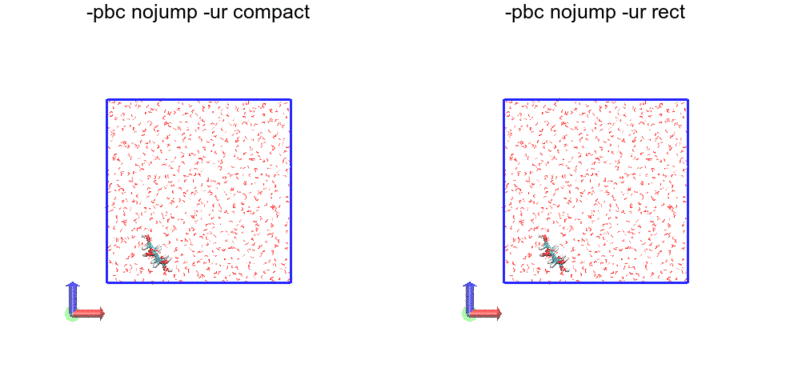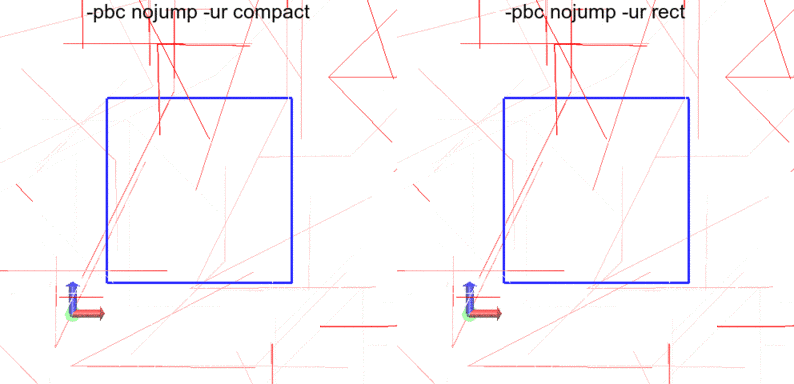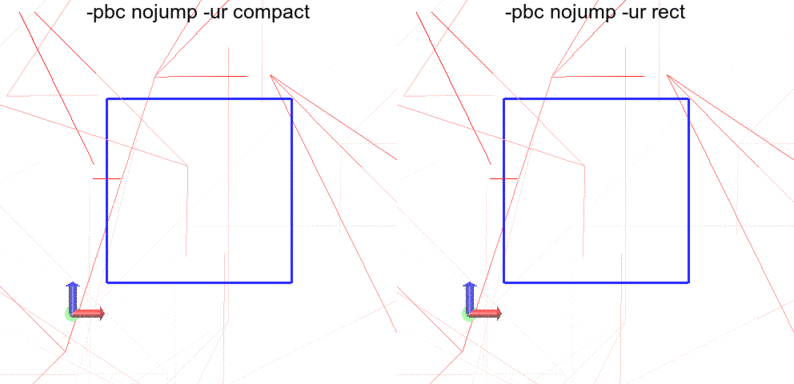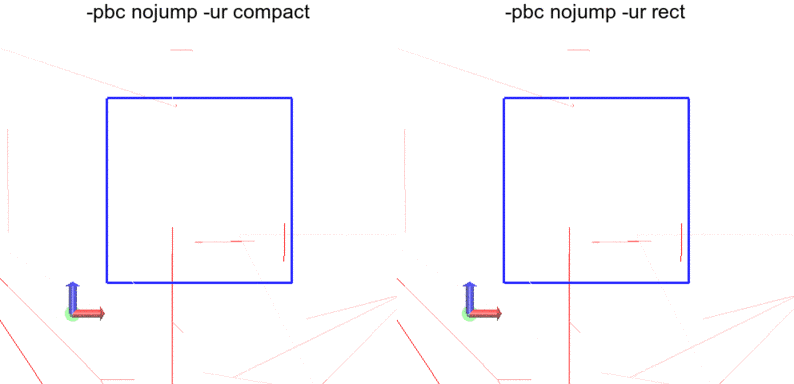
分子がジャンプせず、箱の外に出ていきます。グルコースについては分子のジャンプがなく、うれしいです。しかし、nojumpのはずなのに水分子が途切れたりジャンプしていることに違和感を持つ方もいるかもしれません。
水分子がジャンプする原因は、一言でいうと水の動きが速すぎるためだと考えられます。
目当ての分子がこのように途切れてしまうのを避けるには、トラジェクトリ保存間隔を小さくするか、ボックスサイズを十分大きくすることが必要だと思われます。
詳細な理由の説明には、nojumpのアルゴリズムのイメージが必要です。nojumpは、すべての原子について、前フレームからのxyzいずれかの方向の変位がボックス辺の半分以上である場合に分子が「ジャンプした」とみなし、変位がボックス辺の半分以下になるよう、元に戻します(ボックスサイズのベクトルの適当に整数倍動かせば、どこかで変位が半分以下になります)。フレーム0に対しては、-sで与えた.tprに含まれる座標からの変位を計算するようです。
水の拡散係数は 2.30 × 10-9 m^2s^(-1)などで、これをもとに平均速度を計算すると、今回のトラジェクトリ保存間隔である100 psの間に1.175 nm程度動くようです。ボックスサイズは一辺3 nm程度なので、原子によっては1フレーム(=100 ps)の間にxyzいずれかの方向にボックスの半分以上動いてしまうのはあり得そうです。そして、ある水分子のうち、例えば水素はギリギリボックスの半分以上動いていて、酸素がギリギリ半分以下しか動いてないなら、酸素は-pbc nojumpにより反対側に飛ばされてしまいます(下図)。そうすることで、変位がボックスの半分以下に収まるためです。
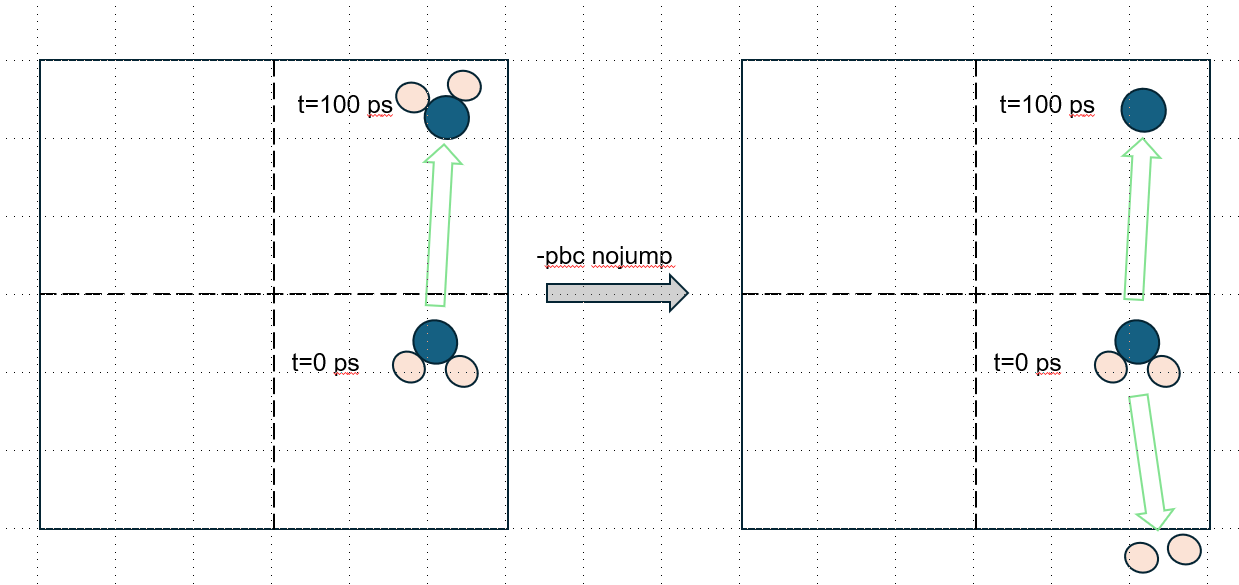
このような場合には、アルゴリズムで「ジャンプのない」トラジェクトリに修正することは不可能だと思われます。したがって、目当ての分子がこのように途切れてしまうのを避けるには、トラジェクトリ保存間隔を小さくするか、ボックスサイズを十分大きくすることが必要です。目安として、1フレームの間に動く距離がボックスの各辺の半分の長さを超えないことが必要です
-pbc nojumpに関する注意
自分がハマッた部分として、2点をここにメモしておきます。
-
-b,-eとの併用時
まず、特定のフレームの切り取りを行う-b,-eオプションと併用する場合、注意が必要です。
別データで試したときに、「前フレームからの変位の確認」は-b,-eで指定した範囲内(+.tpr?)でしか行われないようでした。つまり、最初のフレームからトラジェクトリが連続的になることはありません。
そのため、連続的なトラジェクトリに直しながらフレームの切り取りを行いたい場合は、一度-pbc nojumpを行ってトラジェクトリを変換したのち、再度-b, -eで切り取りを行うという2ステップが必要になります。
-
-sで与えるファイルへの依存性
上の説明でもある通り、ジャンプの判定には-sで与えた.tprも利用されます。したがって、このファイルの構造データ内で分子が非連続的になっている場合、この構造をもとにジャンプしないようにするので、ずっと途切れたままになってしまいます。mdrunの出力する.groをもとにgromppして生成した.tprなら途切れがないので大丈夫ですが、ほかの構造をもとに生成した.tprを用いてnojumpさせる場合は注意が必要です。
3. gmx系の解析ツールのPBC周りの仕様
gmx系の解析ツールの中で、自分の使うことのあったgmx distance, gmx rmsについてです。
まず、gmx distanceですが、-pbc yesだと-pbc molをかけた状態、-pbc noだと-pbc wholeをかけた状態にしてから計算しているようでした。つまり、いずれでも、distanceで計算するグループごとに(あるいは[molecules]ごとに?)PBCを横切らないように修正をかけることはしているようです。デフォルトだとyesです。
参考:ドキュメントより
-[no]pbc (yes) Use periodic boundary conditions for distance calculation
これらの操作では分子の途切れは直せますが、ボックスを超えたジャンプは直せません。もしボックスの外に出てもいいから連続的なトラジェクトリとして分子間の距離や距離ベクトルを計算したい場合は、一度gmx trjconv -pbc nojumpで処理したのちに、gmx distance -pbc noで計算する必要があるでしょう。
次に、gmx rmsですが、こちらにも-pbcオプションがあります。
参考:ドキュメントより
-[no]pbc (yes) PBC check
これら両方のオプションでprd.xtcに対して計算してみます。グルコースでfitしてグルコースでRMSD計算をします。
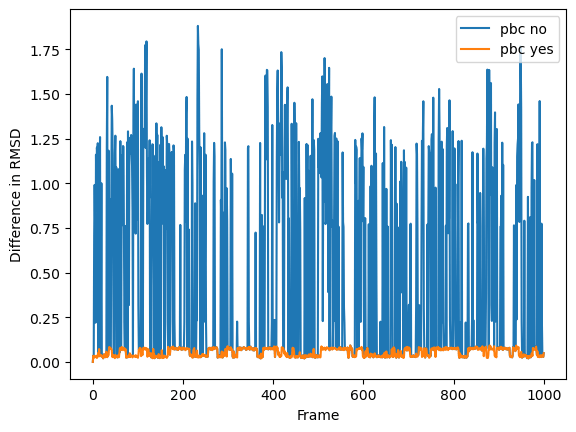
-pbc yesにした場合、rmsdの値が小さく、-pbc noにするとrmsdの値が大きくなっています。大きくなっているのは、途切れた分子をそのままで計算しているからと思われます。distance同様、-pbc yesをつけると途切れている分子の修正を行うようです。ここに関しては、普通にyesでよいような気がします。リガンドrmsdを計算したくて、リガンドがPBCぴょこぴょこしているときなどは少し注意が必要かもしれません。
ちなみに、-sに.tprでなく.groを指定すると、-pbc yesにしても以下のwarningとともに、-pbc noと同じ結果が出ます。pbcの補正は.ndxのgroup単位ではなく、[molecules]単位で行っているようです。補正してほしいなら、.tprを与える必要があります。
WARNING: If there are molecules in the input trajectory file
that are broken across periodic boundaries, they
cannot be made whole (or treated as whole) without
you providing a run input file.
4. mdtrajで解析する場合
.xtcフォーマットは、様々な解析ツールで読み込めます。
自分がよく使ってるmdtrajだと、通常途切れた分子の自動修正は行われません。
最近Trajectoryクラスのドキュメントを見ていて、make_molecules_whole()やimage_molecules()というメソッドがあるのを見つけました。この辺で上のtrjconv -pbcと似たようなことができそうですが、.tprなどのトポロジー情報を読み込めないため、どことどこが同じ分子なのか認識できなそうです。ちょっと試した感じ、タンパク質はある程度トポロジーを自動認識できそうですが、-pbc mol, -pbc wholeに相当する操作をしたい場合、小分子の場合はメソッドの引数としてあらわに指定してあげる必要がありそうでした。すごく小さい分子であれば手動でも何とかなるかもしれませんが、そんな面倒なことをするくらいならgmx trjconvで処理してからmdtrajに渡した方がずっと楽そうです。
ただし、-pbc nojumpに相当する操作はトポロジー情報がいらないので、自分でコードを書くことで再現できる気がします。
5. ユースケース
以上を踏まえて、様々なケースでどのような処理が適切か考えてみます。
実際にうまくいくことを確認したわけではないので、参考程度にしてください。
タンパク質単量体の通常のMD
膜タンパク質などでなければ一般的にタンパク質はだんだんずれていくので、ボックスの境界にまたがり左右に途切れて表示されることが考えられます。このような場合の処理としては以下のようになるでしょう。
- trjcatですべてのトラジェクトリファイルを結合
- (必要なら)trjconvで水分子など解析に不要な部分を除いたトラジェクトリファイルを作成
- (必要なら)gmx convert-tprなどで必要な部分だけを抜き出したtprを作成
- trjconv -pbc molで処理(-pbc nojumpでも可ですが)
- trjconv -fit rot+transで参照構造にfitting(タンパク質でfit)
- gmx, mdtrajなどで解析
タンパク質・リガンド複合体の通常のMD
複合体が境界付近にあるときに-pbc molで処理すると、タンパク質とリガンドが左右に分離されて表示されてしまう可能性があります。このような場合の処理としては以下のようになるでしょう。
ある程度長いトラジェクトリを作る際には、gromppとmdrunを複数回行ってファイルを分けるのが一般的だと思うので、その想定で行きます。
- trjcatですべてのトラジェクトリファイルを結合
- (必要なら)trjconvで水分子など解析に不要な部分を除いたトラジェクトリファイルを作成
- (必要なら)gmx convert-tprなどで必要な部分だけを抜き出したtprを作成
- trjconv -pbc nojump
- trjconv -fit rot+transで参照構造にfitting(タンパク質でfit)
- gmx, mdtrajなどで解析
ただ、水分子も飛び足してしまいます。これがいやな場合は、こちらの記事のような手法が便利で確実だと思います。
分子が箱から飛び出てしまうような細長い分子を視覚上途切れないようにしたい
一部のタンパク質や、合成されたポリマーなど、細長い場合はポリマーの向き次第で変な表示になってしまう場合があると思います。この場合は、上の例と同様に処理することで視覚上は問題ないように変換できると思われます。
でもこの場合、計算時の相互作用としては隣のセルのものが入ってしまっているような気がするので、箱のサイズを大きくするのが適切かもしれません。
スナップショットの選択・計算の再開を繰り返すようなサンプリング
ちょっと複雑になります。
- スナップショットを取り出す際は、
trjconv -pbc nojumpによる処理、-b,-eによる切り出しの処理の2ステップで行う - 各MDのデータに対して、
trjconv -pbc nojumpで処理。-sには該当MDの計算に利用した.tprを使う - trjconv -fit rot+transで参照構造にfitting(タンパク質でfit)
- gmx, mdtrajなどで解析
このパターンについてのみ、例を載せておきます。スナップショットの選択・リスタートを繰り返すサンプリング手法であるPaCS-MDにおいて、グルコースと水分子の解離シミュレーションを行った場合の例になります。(あくまで例なので、実際はこんな変な解離シミュレーションはしないと思います。)
PaCS-Toolkitでnojump=trueとして計算を行うと、解離していく水分子について、このような軌跡を得ることができます。
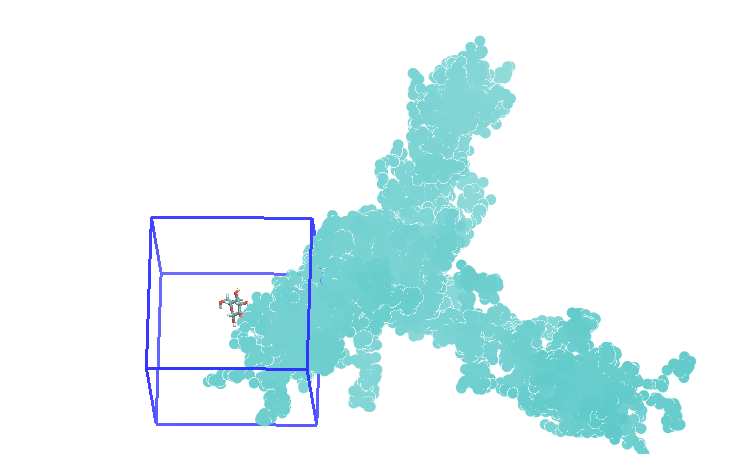
この例ではすべてのスナップショットの重心を可視化しています。ボックスをはみ出してもそのままトラジェクトリが連続していることがわかります。
もっとも、これぐらい大きくボックスからはみ出てしまうと相互作用については問題が出てくると思います。nojumpオプションは、あくまで、少しだけリガンドがボックスをはみ出す場合にartifactを生まないための修正方法として提供されています。
余談:.mdpのオプション
.mdpで与えるオプションとして、pbc, periodic_moleculesのオプションがありますが、これらは計算時の設定なので、今回対象とする視覚化や解析時の話とはあまり関係がなさそうです。
おわりに
gromacs、特にtrjconvコマンドのオプションを中心に、PBCの取り扱いについてまとめました。
もし間違っているところなどございましたらご指摘いただければ大変ありがたいです。
Discussion