分子解析やデザインに使えるPyMOLコマンドのextentions
PyMOLは分子解析ツールなのでタンパクや核酸の構造を解析するだけでなく、低分子や抗体(多くはFab?)との複合体構造の解析が創薬の一助となります。
メディシナルケミストの目線ではやはり低分子とその周辺残基の解析が最も重要です。
多くの企業は有償のソフトウェアを導入していると思うのであまり必要はないかと思いますが、計算をあまり投げずに分子解析をするだけなら動作が軽量なPyMOLは非常に優秀です。
ここでは解析にあたってケミスト目線で使うコマンドのextensionのスクリプトを公開します。
バックグラウンドがケミストなのでコーディングの面で難しい内容はありませんが、PyMOLにインストールすると結構便利です。ご参考までに。
residues.py
説明は後述しますが、ひとまず下記のスクリプトをresidues.pyとして保存して、PyMOLを起動した後に実行しておくと新しいコマンドが使えるようになります。
from pymol import cmd
# Visualization
### One-letter Labeling
one_letter ={'VAL':'V', 'ILE':'I', 'LEU':'L', 'GLU':'E', 'GLN':'Q', \
'ASP':'D', 'ASN':'N', 'HIS':'H', 'TRP':'W', 'PHE':'F', 'TYR':'Y', \
'ARG':'R', 'LYS':'K', 'SER':'S', 'THR':'T', 'MET':'M', 'ALA':'A', \
'GLY':'G', 'PRO':'P', 'CYS':'C'}
cmd.alias('tag', 'label n. CA and sele, "%s%s" % (one_letter[resn], resi)')
cmd.alias('b', 'label n. CA and sele, b')
cmd.alias('untag', 'hide label, sele')
### hide sticks, show residues
cmd.alias('k', 'hide sticks, polymer.protein; show sticks, enabled and resn lys')
cmd.alias('c', 'hide sticks, polymer.protein; show sticks, enabled and resn cys')
cmd.alias('s', 'hide sticks, polymer.protein; show sticks, enabled and resn ser')
cmd.alias('h', 'hide sticks, polymer.protein; show sticks, enabled and resn his')
cmd.alias('t', 'hide sticks, polymer.protein; show sticks, enabled and resn thr')
cmd.alias('r', 'hide sticks, polymer.protein; show sticks, enabled and resn arg')
cmd.alias('d', 'hide sticks, polymer.protein; show sticks, enabled and resn asp')
cmd.alias('e', 'hide sticks, polymer.protein; show sticks, enabled and resn glu')
cmd.alias('n', 'hide sticks, polymer.protein; show sticks, enabled and resn asn')
cmd.alias('q', 'hide sticks, polymer.protein; show sticks, enabled and resn gln')
cmd.alias('y', 'hide sticks, polymer.protein; show sticks, enabled and resn tyr')
cmd.alias('f', 'hide sticks, polymer.protein; show sticks, enabled and resn phe')
cmd.alias('w', 'hide sticks, polymer.protein; show sticks, enabled and resn trp')
cmd.alias('g', 'hide sticks, polymer.protein; show sticks, enabled and resn gly')
cmd.alias('a', 'hide sticks, polymer.protein; show sticks, enabled and resn ala')
cmd.alias('m', 'hide sticks, polymer.protein; show sticks, enabled and resn met')
cmd.alias('p', 'hide sticks, polymer.protein; show sticks, enabled and resn pro')
cmd.alias('i', 'hide sticks, polymer.protein; show sticks, enabled and resn ile')
cmd.alias('l', 'hide sticks, polymer.protein; show sticks, enabled and resn leu')
cmd.alias('v', 'hide sticks, polymer.protein; show sticks, enabled and resn val')
cmd.alias('me', 'hide sticks, polymer.protein; show sticks, enabled and resn val; show sticks, enabled and resn leu; show sticks, enabled and resn ile; show sticks, enabled and resn met')
cmd.alias('z', 'hide sticks, polymer.protein')
cmd.alias('addh', 'cmd.h_add("sele")')
cmd.alias('remh', 'cmd.remove("sele extend 1 & hydro & not nbr. (don.|acc.)")')
### water and hydrophobic hydrogens manupilations
cmd.alias('del', 'as nb_spheres, solvent; hide nb_spheres')
cmd.alias('sol', 'as nb_spheres, solvent')
cmd.alias('ph', 'hide (hydro) and enabled and (elem C extend 1)')
### coloring
def white():
util.cba(144, "enabled", _self=cmd)
cmd.extend('white',white)
def cc():
util.color_chains("(enabled and name CA)", _self=cmd)
cmd.extend('cc',cc)
def dssp():
cmd.color('gray', 'enabled and ss l')
cmd.color('yellow', 'enabled and ss s')
cmd.color('red', 'enabled and ss h')
cmd.extend('dssp',dssp)
def plddt():
cmd.color('0x0053D6', 'enabled and b < 100')
cmd.color('0x65CBF3', 'enabled and b < 90')
cmd.color('0xFFDB13', 'enabled and b < 70')
cmd.color('0xFF7D45', 'enabled and b < 50')
util.cnc()
cmd.color('white', 'enabled and sc. and elem C')
cmd.extend('plddt',plddt)
## interaction analysis
### zoom into around ligand
def see(distance=8):
cmd.zoom('byres organic around %s and enabled' % distance)
cmd.extend('see', see)
### show lines around ligand
def line(distance=4):
cmd.show('lines', 'byres organic around %s and enabled' % distance)
cmd.extend('line', line)
### select residues with distance around ligand
def grab(sele='organic', distance=4):
cmd.select('sele', 'byres %s around %s and enabled' % (sele, distance))
cmd.enable('sele')
cmd.extend('grab', grab)
### show polar contact of ligand to residues in the enabled same object
def hb():
cmd.select('ligand', 'organic and enabled')
cmd.dist('contact', '(ligand)', '(byobj (ligand)) and (not(ligand))', quiet=1, mode=2, label=0, reset=1)
cmd.enable('contact')
cmd.extend('hb',hb)
### show polar contact of ligand to residues between the enabled different object
def dock(ligand='enabled and organic'):
cmd.delete('polar_conts')
cmd.dist("polar_conts","%s" % ligand, "(not %s)" % ligand ,quiet=1,mode=2,label=0,reset=1)
cmd.enable("polar_conts")
cmd.extend('dock', dock)
### show polar contact of sele to residues
def at():
cmd.dist('contact', '(sele)', '(byobj (sele)) and (not(sele))', quiet=1, mode=2, label=0, reset=1)
cmd.enable('contact')
see(distance)
cmd.extend('at',at)
### create object of ligand and residues with distance
def byres(distance=8, ligand='organic and enabled'):
object_name = 'byres' + str(distance)
cmd.create('Lig', '%s' % ligand)
cmd.create(object_name, 'byres %s around %s and enabled' % (ligand, distance))
cmd.disable('!' + object_name)
cmd.enable('Lig')
line()
dock('Lig')
see()
grab()
cmd.extend('byres',byres)
## sequencing
### print fasta sequence of enabled object
def fasta():
print(cmd.get_fastastr('enabled'))
cmd.extend('fasta',fasta)
### print fasta sequence of sele object
def seq():
print(cmd.get_fastastr('sele'))
cmd.extend('seq',seq)
上記のスクリプトをresidues.pyとして保存したディレクトリで下記のコマンドをPyMOLのコマンドラインで実行しておきます。
run ./residues.py
これで準備は完了です。
使い方
スクリプト記載の順番ではありませんが、ご容赦ください。
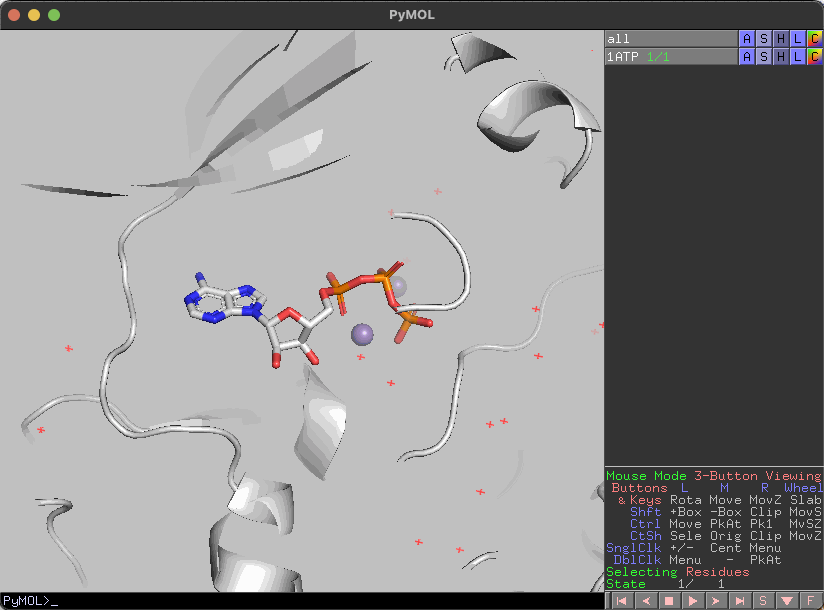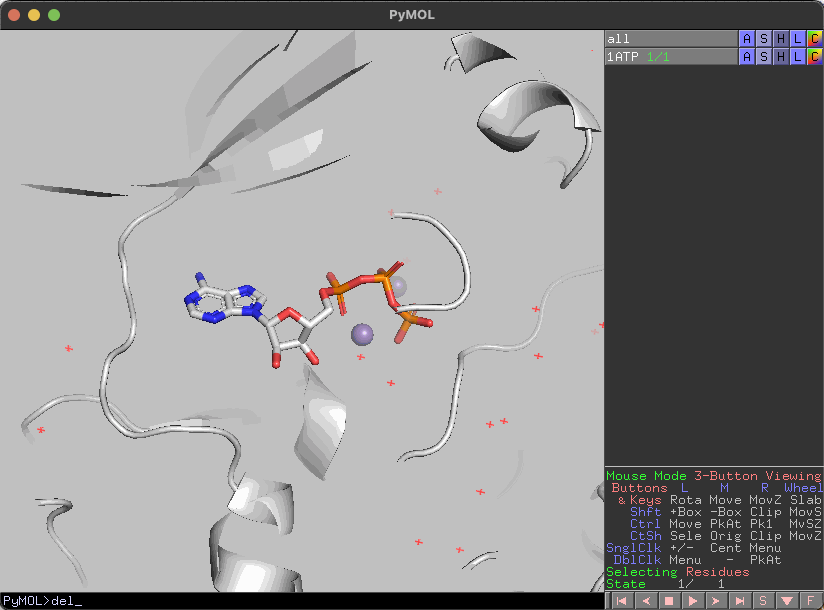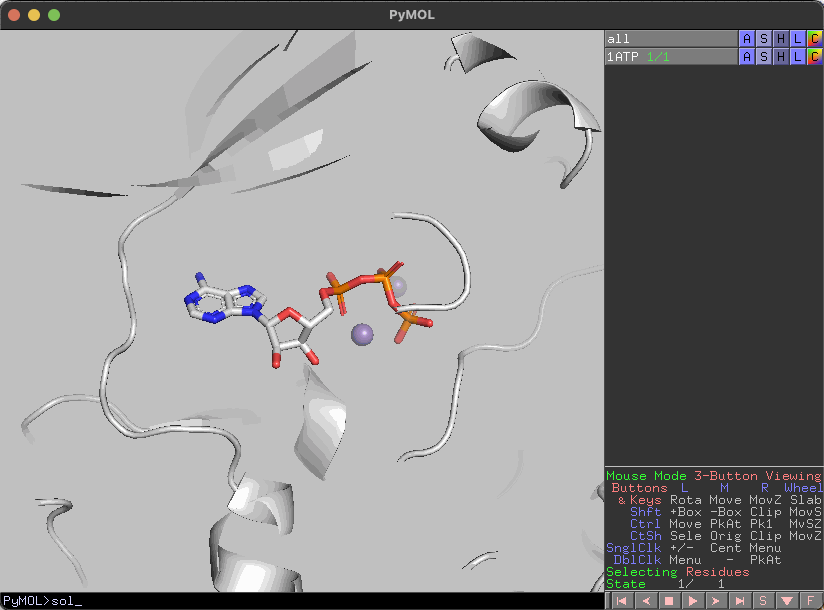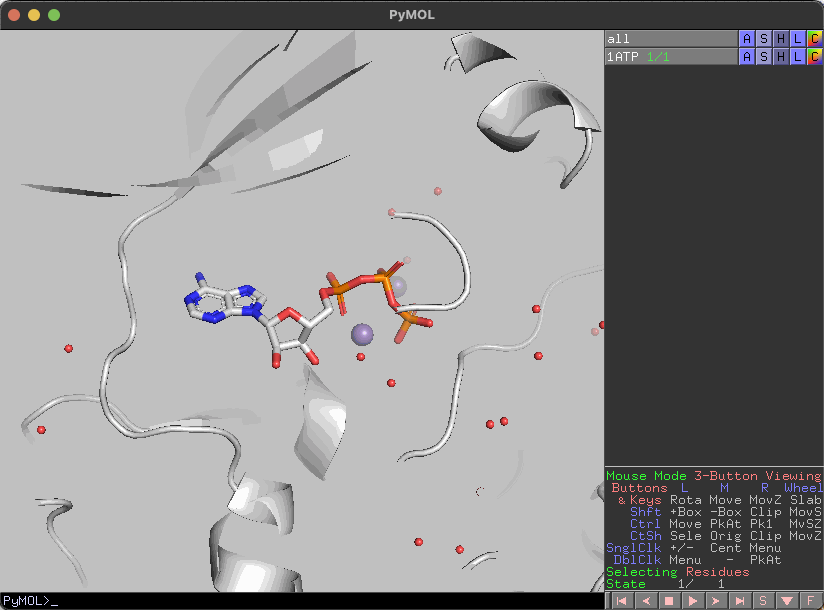
水分子の表示/非表示
水分子を非表示にしたい場合はdel、表示したい場合はsolを実行すると水分子の表示を切り替えることができます。
del
sol
基本的には全objectに対して実行されるはずです。object指定は気が向いたら対応します(え?
水素原子の付加/除去
ph
表示しているオブジェクトについて、非極性水素を非表示にします。
addh
選択している分子に擬似的に水素原子を付加します。
構造解析では通常、分子の水素原子が観測されていません。
確かβ線を用いる測定法では最外殻に電子のない水素は観測できない、みたいな理由だった気がします。
中性子線を用いる測定では水素も観測できたんじゃなかったでしょうか。
水素原子を付加できると、簡易的に原子間距離や角度を測ったり、builderで水素から別の原子に置換して分子デザインすることができるようになります。
注)実はPyMOLにはデフォルトでh_addという同じコマンドが存在してました。。勉強不足でごめんなさい🙇
反対に、水素原子を除去する場合は下記のコマンドです。
remh
書いておいてなんですが、remhはあまり使った試しはありません。。
水素原子を非表示にするphとの違いとして、remhは構造から水素原子を除去しています。
どちらもsurfaceに影響するので注意が必要です。
特定のアミノ酸をstick表示する
いわゆるone-lette表示のスクリプトを書いてあるので、アミノ酸を入力実行すると指定した残基をstick表示します。
注意点としては、他のstick表示したアミノ酸は非表示になります。ただし、linesやspheresは影響を受けません。
また、zで全てのアミノ酸を非表示にします。
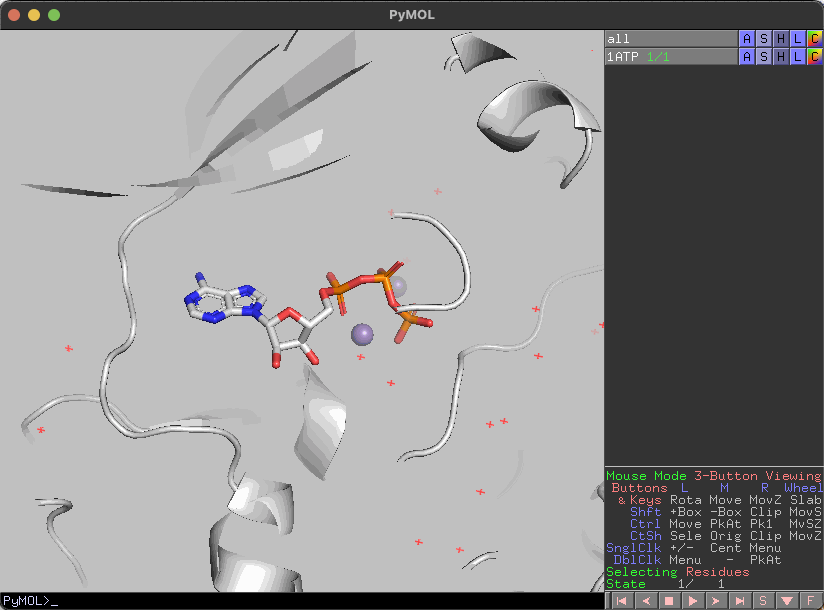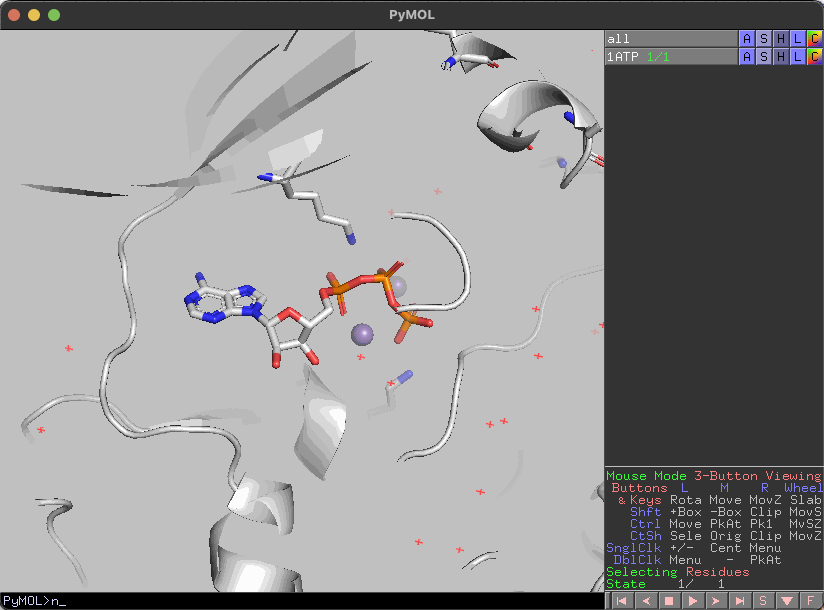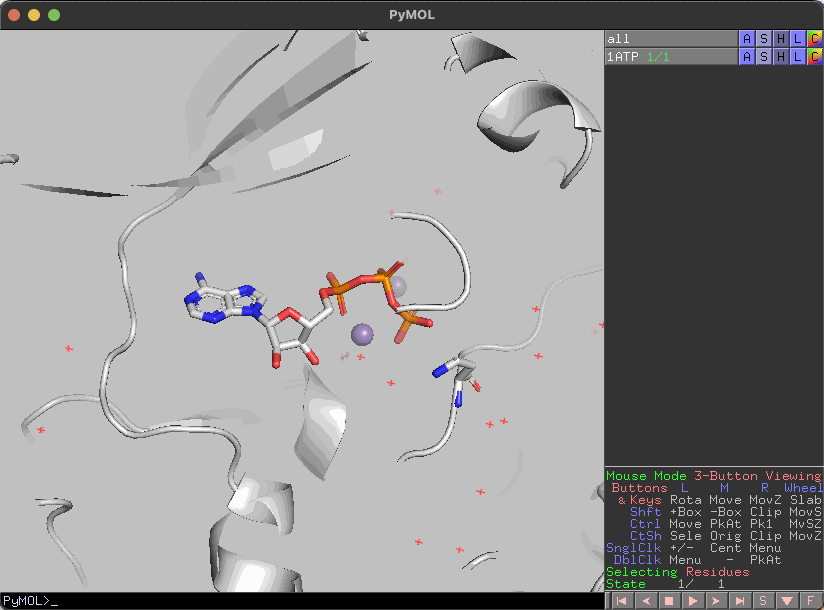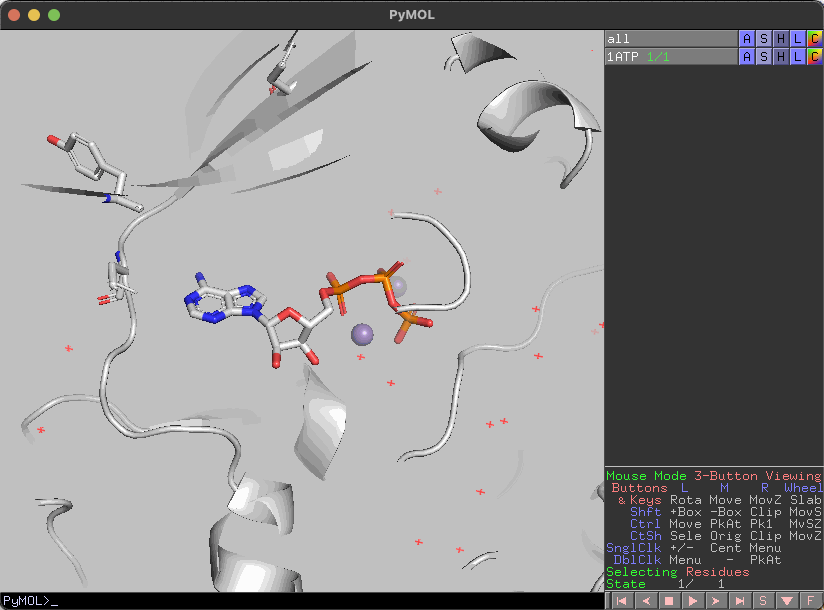
選択したアミノ酸のラベルを表示する
tag
(sele)のオブジェクトのアミノ酸残基のラベルを表示します。
untag
tagとは反対に、(sele)のオブジェクトのアミノ酸残基のラベルを非表示にします。
b
(sele)のオブジェクトのアミノ酸残基のb factorを表示します。
カラーリング
cc
こちらの記事でも紹介しましたが、color by chainに準拠しています。
white
バックボーンを白に変換します。
酸素は赤、窒素は青、塩素は緑、リンはオレンジのように、color by elementのwhiteに(確か)準拠しています。
dssp
DSSP(水素結合推定アルゴリズム)というアルゴリズムに基づいた二次構造を元にカラーリングしています。
PyMOLのabbreviationでいうssに相当してます。
plddt
AlphaFold2の構造をloadすると、予測スコアのplddtがb factorに格納されています。
下記の記事を参考にカラーリングしつつ、側鎖の配色がうざったかったので炭素はwhiteに設定しています。
相互作用解析
前提として、私自身がメドケムなので、その視点でのコマンドを作っています。
構造生物やde novo designの方は、また違った観点で解析してるんじゃないかなと思います。
例えば、私自身は電子密度のマップやb factorよりも、特に低分子周辺の水素結合ネットワークをよく観察することが多いです。
構造情報の精密性よりも分子デザインのための解釈を重要視しているからです。
以下が大体の流れかなと思います。
see
有機分子の周辺8Åにzoomします。
有機分子がない場合は表示しているobjectの重心周辺8Åです(多分)。
line
有機分子周辺4Åにあるアミノ酸残機のlinesを表示します。
grab
lineで表示されるアミノ酸残基で(sele)オブジェクトを作ります。
そのままtagを使うとラベルを表示できます。
byres [distance=8, ligand='organic and ebabled']
byresは引数を指定しない場合、表示しているオブジェクトから有機分子周辺8Åについて新しいオブジェクトを作ります。
その後、lineとgrabで処理された状態になります。tagまでは処理していません。
水分子はついてくるはずです。
byres 12
上記のように第一引数に数字を含めると、新しいオブジェクトに含めるアミノ酸残基の範囲を変更できます。
hb
オブジェクト内の有機分子からアミノ酸残基や水分子に対しての極性相互作用を表示します。
dock
表示されているオブジェクト内の有機分子からアミノ酸残基や水分子に対しての極性相互作用を表示します。
hbとの違いはオブジェクトを越えて表示できることです。
機会があれば紹介したいですが、例えばdockingで得られたposeに対して相互作用を解析できます。
at
(sele)のオブジェクトから周辺4Åのアミノ酸残機や水分子に対する極性相互作用を表示します。
配列情報
fasta
表示している配列のFASTAをPyMOLのExternal GUIに出力します。
seq
(sele)のオブジェクトのFASTAをPyMOLのExternal GUIに出力します。
終わりに
多くの方はGUIの操作に慣れていると思いますが、コマンドで色んな処理が出来るようになると解析の速度が速くなります。
PyMOLに慣れてきた方は参考にしてみてください。
Discussion