プログラム、下から作るか?上から作るか?
TL;DR
- プログラムは「下から組む方法」と「上から組む方法」がある
- プログラムを組む時は少しずつテストしながら組む
はじめに
なにかゼロからプログラムを組むとします。そのプログラムのアルゴリズムや、何をやるべきかはなんとなくわかっているけれど、どこから手をつけてよいかがわからず、ChatGPTに全部書かせて、その後修正できずに困る、という事例を何度か観測しています。
プログラムをゼロから書くのは慣れが必要です。プログラムをゼロから書く場合、小さな部品を一つ一つ作っていって、最後にそれらを組み上げる「下から書く」方法と、「こういう関数が必要であるはず」と外枠から書いていって最後に中身を埋める「上から書く」方法があります。その一般論を論じるのは私の能力を超えるため、以下では「下から」と「上から」の例を挙げて、その「気持ち」を説明してみようと思います。言語はなんでも良いですが、ここではPythonを使います。
下から書く例「パーコレーション」
Union-Find
正方格子が与えられたとします。その格子のボンドが確率
各格子点をノードと呼び、それらに通し番号をつけ、アクティブなボンドで接続されたノードは同じクラスターであるとします。この時、「友達の友達は友達」と定義し、ノードAとBが同じクラスター、BとCが同じクラスターなら、AとCも同じクラスターであると定義します。ノードの集合と、アクティブなボンドでつながったノードペアの集合が与えられた時、任意の2つのノードが同じクラスターに属すかどうかを調べる手段を提供するのがUnion-Findアルゴリズムです。
Union-Findアルゴリズムは、2つのノードをつなげるunion関数と、あるノードのクラスター番号を返すfind関数の2つからなります。いくつか実装がありますが、一番簡単な一次元配列を使う実装にしましょう。では、コードを書き始めます。
main関数の作成
最初にやることは、以下のようにmain関数を作ることです。
def main():
pass
if __name__ == "__main__":
main()
if __name__ == はPythonのイディオムで、このスクリプトを直接実行した時のみ実行されるようにするものです(他のスクリプトからimportされた時は実行されない)。ここからmain関数を呼び、あとはそこに処理を追加していきます。このようにmain関数を作る意味はグローバル変数を使わないようにするためです。グローバル変数はあとでバグの温床になります。プログラムの起点を関数にしておき、必要な変数を関数内で定義しておくと、「必要な変数は引数として渡す」という癖がつきます。
このifの中にテストコードを書いてはいけません。Pythonのif文はスコープを作らないため、ここに書いた変数はグローバル変数になります。mainでもtestでもどんな名前でもかまいませんが、スタート地点は関数にしておく必要があります。
あとは、関数を追加しては、main関数にテストコードを書いて呼び出す、という手順を繰り返します。
find関数の作成
最初に作るのはfind関数です。自分の「親」をたどっていって、一番上に到達したらその番号を返す、というコードです。インデックス(index)と一次元配列(cluter)を受け取って、whileを回すだけの簡単な関数です。こんな感じのコードになるでしょうか(わざと間違えています)。
def find(index, cluster):
while index != cluster[index]:
index = cluster[index]
return index
このコードを試してみましょう。テストコードをmainに書きます。5つノードがあり、
def main():
cluster = [0, 0, 1, 2, 3]
for i in range(len(cluster)):
print(i, find(i, cluster))
上記の配列は、以下のようなノード接続状況を表しています。
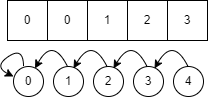
つまり、すべてが同じクラスターに所属し、ルートノードは0です。従って、想定される出力は、すべてのクラスター番号が0と表示されるものです。しかし、実行してみるとこうなります。
0 None
1 0
2 1
3 2
4 3
想定出力と異なります。ここでfind関数を見ると、return文のインデントを間違えていることに気づきます。直しましょう。
def find(index, cluster):
while index != cluster[index]:
index = cluster[index]
return index # ここのインデントがおかしかった。
もう一度実行します。
0 0
1 0
2 0
3 0
4 0
想定通りの出力になりました。まだコーナーケースのバグは残っているかもしれませんが、とりあえずfind関数は完成です。こうやって、一つ関数を書くたびに、必ず簡単なケースでテストします。
union関数の作成
次にunionを作ります。2つのノードをつなぐのが目的です。それぞれのノード番号を受け取り、findを使ってクラスター番号を受け取って、小さい方のクラスター番号に揃えます。これも簡単な関数です。
def union(index1, index2, cluster):
c1 = find(index1, cluster)
c2 = find(index2, cluster)
if c1 < c2:
cluster[c2] = c1
else:
cluster[c1] = c2
試してみましょう。5つノードがあり、1と2、3と4、最後に1と4をつなぎます。すると、0以外のノードがすべてクラスター番号1になるはずです。
def main():
N = 5
cluster = [i for i in range(N)]
union(1, 2, cluster)
union(3, 4, cluster)
union(1, 4, cluster)
for i in range(N):
print(i, find(i, cluster))
実行してみましょう。
0 0
1 1
2 1
3 1
4 1
想定通りの出力になりました。
show関数の作成
これからのデバッグのため、すべてのノードのクラスター番号を表示する関数を作っておきます。乱数を使うようなコードは、特にデバッグに気を使わなくてはいけません。システムサイズをcluster配列を受け取って、その中身を表示する関数showを作っておきましょう。
def show(L, cluster):
for iy in range(L):
for ix in range(L):
index = ix + iy * L
c = find(index, cluster)
print(f"{c:02d} ", end="")
print()
デバッグのため、とりあえずクラスター番号は2桁で止めておきます。
clusterを初期化しておいて、showを呼ぶテストを書きましょう。
def main():
L = 5
N = L * L
cluster = [i for i in range(N)]
show(L, cluster)
ここで、show(5, cluster)のように、直接サイズを渡さず、変数経由で渡しています。関数の引数の生の数字を入れてはいけません。なるべく変数経由で渡す癖をつけておきましょう。
実行してみます。
00 01 02 03 04
05 06 07 08 09
10 11 12 13 14
15 16 17 18 19
20 21 22 23 24
まだどのノードも接続していないため、すべてのノードのクラスター番号が異なる状態です。
mc_onestep関数の作成
次に、確率mc_onestepという名前にしましょうか。これはサイズと確率
そのためには、座標を2つ受け取り、確率connect関数も必要です。これらは同時に作りましょう。
まず、connect関数はこんな感じになるでしょう。デバッグ用にどことどこをつないだか表示しておきます。
def connect(p, x1, y1, x2, y2, L, cluster):
i1 = x1 + y1 * L
i2 = x2 + y2 * L
if random.random() < p:
union(i1, i2, cluster)
print(f"Connect: {i1} {i2}")
connectを呼ぶ、mc_onestep関数はこうなるでしょう。
def mc_onestep(p, L):
L = 5
N = L * L
cluster = [i for i in range(N)]
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix + 1, iy, L, cluster) # (1)
show(L, cluster)
本当は、# (1)の部分はconnect(p, ix, iy, ix, iy + 1, L, cluster)と、y方向につなぐ必要がありますが、コピペして修正し忘れた、という想定で間違えています。また、最後にデバッグ用にshowを呼んでいます。
また、ここには書いていませんが、コードの冒頭にimport randomを追加します。
これらをテストするmain関数を書きます。まずは確率0.5でためしましょう。
def main():
random.seed(0)
L = 5
p = 0.5
mc_onestep(p, L)
ここでrandom.seed(0)を指定していることに注意してください。乱数を使うコードは、デバッグ時は必ずシードを固定します。そうしないと「あれ?」と思った時に状況が再現できずに困るからです。デバッグの前提は、同じ状況で同じ結果が出ることです。そこが崩れるとデバッグが極めて困難になります。サーバ系や並列コードなど、非同期な処理のデバッグが難しいのは、全く同じ状況を再現することが難しいことによります。
さて、実行しましょう。
Connect: 5 6
Connect: 5 6
Connect: 10 11
Connect: 15 16
Connect: 1 2
Connect: 11 12
Connect: 16 17
Connect: 12 13
Connect: 3 4
Connect: 3 4
Connect: 8 9
Connect: 18 19
00 01 01 03 03
05 05 07 08 08
10 10 10 10 14
15 15 15 18 18
20 21 22 23 24
必ず2回実行し、同じ結果が得られることを確認します。乱数系のコードは、例えばnumpyやtensorflowは独自のシードを持っていたりするため、シードを固定しても結果が変わる場合があります。「毎回同じ結果になる」ことを確認してから次に進みます。
さて、このコードを見て「あっ、y方向の接続を忘れた」と気づくのは難しいでしょう。ですが、
def main():
random.seed(0)
L = 5
p = 1.0
mc_onestep(p, L)
すべてのボンドがアクティブなのだから、すべてのノードがつながっているはずです。実行してみましょう。
00 00 00 00 00
05 05 05 05 05
10 10 10 10 10
15 15 15 15 15
20 21 22 23 24
これを見ると、x方向だけが接続され、y方向の接続を忘れているのが一目瞭然です。修正しましょう。
def mc_onestep(p, L):
L = 5
N = L * L
cluster = [i for i in range(N)]
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster) # ここを修正
show(L, cluster)
改めて実行してみます。
00 00 00 00 00
00 00 00 00 00
00 00 00 00 00
00 00 00 00 00
00 00 00 00 24
右下が接続されていません。これは、ループをrange(L-1)で回したため、右端と下端の接続を忘れていたためです(すみません、これはわざとではなく素で忘れました)。
右端と下端の接続を追加し、それ以外の接続をなしにして、ちゃんと繋がるか確認しましょう。
まずは右端のみの確認です。
def mc_onestep(p, L):
L = 5
N = L * L
cluster = [i for i in range(N)]
"""
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster)
"""
for iy in range(L - 1):
connect(p, L - 1, iy, L - 1, iy + 1, L, cluster)
show(L, cluster)
実行します。
00 01 02 03 04
05 06 07 08 04
10 11 12 13 04
15 16 17 18 04
20 21 22 23 04
右端が繋がりましたね。同様に下端も確認します。
def mc_onestep(p, L):
L = 5
N = L * L
cluster = [i for i in range(N)]
"""
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster)
for iy in range(L - 1):
connect(p, L - 1, iy, L - 1, iy + 1, L, cluster)
"""
for ix in range(L - 1):
connect(p, ix, L - 1, ix + 1, L - 1, L, cluster)
show(L, cluster)
00 01 02 03 04
05 06 07 08 09
10 11 12 13 14
15 16 17 18 19
20 20 20 20 20
下端が繋がりました。改めて全部をつなげてみましょう。
def mc_onestep(p, L):
L = 5
N = L * L
cluster = [i for i in range(N)]
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster)
for iy in range(L - 1):
connect(p, L - 1, iy, L - 1, iy + 1, L, cluster)
for ix in range(L - 1):
connect(p, ix, L - 1, ix + 1, L - 1, L, cluster)
show(L, cluster)
実行します。
00 00 00 00 00
00 00 00 00 00
00 00 00 00 00
00 00 00 00 00
00 00 00 00 00
ちゃんと繋がりました。
適当な確率、例えば
Connect: 5 6
Connect: 5 10
Connect: 10 15
Connect: 15 20
Connect: 1 2
Connect: 11 12
Connect: 16 21
Connect: 12 13
Connect: 3 4
Connect: 3 8
Connect: 8 9
Connect: 18 19
Connect: 4 9
Connect: 19 24
Connect: 21 22
00 01 01 03 03
05 05 07 03 03
05 11 11 11 14
05 16 17 18 18
05 16 16 23 18
ちゃんとノートに図を書いて、どことどこがつながったからこのクラスターになるはず、ということを確認しましょう。
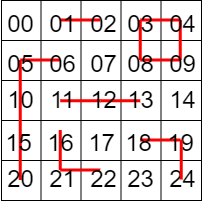
「5と6がつながって、10と15がつながって・・・」と真面目に書きます。こういうところで手を抜かないのがプログラミングのコツです。できた図を先程のクラスタリング結果と比較して、間違いないことを確認します。
ここまで大丈夫そうなので次に行きます。union関数のデバッグ表示は消しておきましょう。
percolation_check関数の作成
クラスタリングが終了したら、上辺と下辺がつながっていることを確認します。もっと効率的なコードはありますが、まずはナイーブ組みましょう。上辺と下辺のノードのクラスター番号が一致しているかどうか調べ、一致していたらTrueを、そうでなければFalseを返す関数です。こんな感じになるでしょうか。
def percolation_check(L, cluster):
for ix1 in range(L):
c1 = find(ix1, cluster)
for ix2 in range(L):
c2 = find(ix2 + (L - 1) * L, cluster)
if c1 == c2:
return True
return False
これをmc_onestepから呼び出します。
def mc_onestep(p, L):
N = L * L
cluster = [i for i in range(N)]
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster)
for iy in range(L - 1):
connect(p, L - 1, iy, L - 1, iy + 1, L, cluster)
for ix in range(L - 1):
connect(p, ix, L - 1, ix + 1, L - 1, L, cluster)
show(L, cluster)
if percolation_check(L, cluster):
print("Percolated")
else:
print("Not Percolated")
main関数から10回ほど実行してみましょう。
def main():
random.seed(0)
L = 5
p = 0.5
for _ in range(10):
mc_onestep(p, L)
実行してみます。
00 01 01 03 03
05 05 07 03 03
05 11 11 11 14
05 16 17 18 18
05 16 16 23 18
Not Percolated
00 00 00 00 04
00 00 07 00 09
00 00 00 00 00
00 00 17 00 00
20 21 22 00 00
Percolated
00 01 02 03 03
05 06 02 03 03
05 02 02 03 14
05 16 03 03 03
20 21 21 23 24
Not Percolated
00 00 00 00 00
00 00 00 00 00
00 00 00 00 14
00 00 00 00 19
00 00 19 19 19
Percolated
00 00 02 02 02
00 00 02 02 02
00 00 00 02 02
00 00 00 02 02
00 00 00 02 02
Percolated
00 00 00 03 04
00 00 00 03 04
10 10 12 12 12
15 12 12 12 12
12 12 12 12 12
Not Percolated
00 01 01 03 03
01 01 07 08 08
10 11 12 08 08
10 10 17 17 08
10 10 10 17 17
Not Percolated
00 00 00 03 04
00 00 00 00 00
00 00 12 00 00
00 00 00 00 00
00 00 00 00 24
Percolated
00 00 00 00 00
00 00 00 00 09
00 00 00 00 09
15 00 17 00 00
00 00 17 00 00
Percolated
00 00 00 00 04
00 00 00 00 09
10 00 00 13 14
15 00 00 18 19
20 21 21 18 19
Not Percolated
上から下までつながっている時にPercolated、そうで無いときにNot Percolatedと表示されていますね。
mc_average関数の作成
次にmc_onestepを何度も呼んで、パーコレーション確率を計算する関数mc_averageを書きましょう。サンプル回数を受け取り、その回数だけmc_onestepを呼び出して、平均を返すだけです。
まず、mc_onestep関数を、パーコレートしていたら1.0を、そうでなければ0.0を返すように修正します。デバッグ情報のshowも削除しておきます。
def mc_onestep(p, L):
N = L * L
cluster = [i for i in range(N)]
for ix in range(L - 1):
for iy in range(L - 1):
connect(p, ix, iy, ix + 1, iy, L, cluster)
connect(p, ix, iy, ix, iy + 1, L, cluster)
for iy in range(L - 1):
connect(p, L - 1, iy, L - 1, iy + 1, L, cluster)
for ix in range(L - 1):
connect(p, ix, L - 1, ix + 1, L - 1, L, cluster)
if percolation_check(L, cluster):
return 1.0
else:
return 0.0
mc_averageは、確率p、サンプル数num_samples、システムサイズLを受け取り、その確率
def mc_average(p, num_samples, L):
sum = 0.0
for _ in range(num_samples):
sum += mc_onestep(p, L)
sum /= num_samples
return sum
このあたりは難しくないので、ミスも少ないでしょう。
main関数から呼び出します。
def main():
random.seed(0)
L = 5
p = 0.5
num_samples = 100
print(mc_average(p, num_samples, L))
実行しましょう。
0.58
この時、p=0のときに0になることと、p=1の時に1.0になることを必ず確認する癖をつけておきましょう。乱数を使ったコードのデバッグは大変ですが、パラメータの極限では厳密に値がわかっていることが多いです。例えばスピン系のモンテカルロ法なら、高温極限と低温極限は必ず確認する癖をつけるようにします。
mc_all関数の作成
最後に、様々なmc_allを作ります。受け取る引数はシステムサイズとサンプル数で良いでしょう。
def mc_all(L, num_samples):
ND = 20
for i in range(ND + 1):
p = i / ND
print(f"{p} {mc_average(p, num_samples,L)}")
確率NDとしていますが、あとで必要になったら引数で渡すように修正しましょう。
mainから呼び出します。
def main():
random.seed(0)
L = 5
num_samples = 100
mc_all(L, num_samples)
実行しましょう。
0.0 0.0
0.05 0.0
0.1 0.0
0.15 0.0
0.2 0.01
0.25 0.06
0.3 0.1
0.35 0.22
0.4 0.34
0.45 0.38
0.5 0.61
0.55 0.82
0.6 0.84
0.65 0.92
0.7 0.93
0.75 0.99
0.8 1.0
0.85 1.0
0.9 1.0
0.95 1.0
1.0 1.0
できてそうですね。サイズとサンプル数を増やして、プロットしてみましょう。L=16の例です。
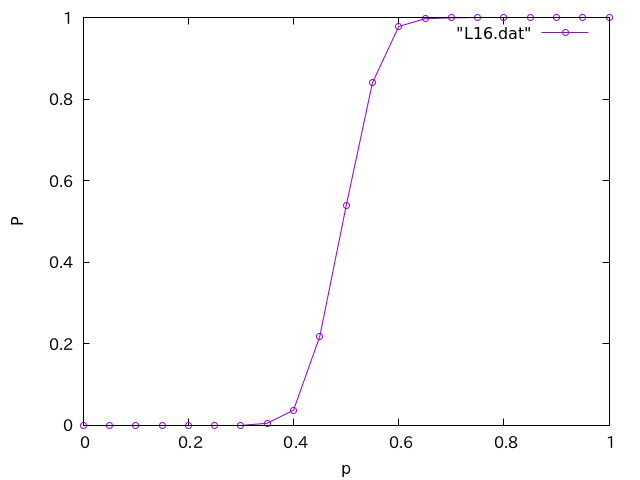
相転移していそうです。
最後に、結果を標準出力に吐くのではなく、システムサイズによってファイル名を決め、そこに出力するように修正しておきましょう。また、実行時引数としてサイズを受け取るように修正します。なお、コードの冒頭にimport sysが必要です。
def mc_all(L, num_samples):
ND = 20
filename = f"L{L:02d}.dat"
print(filename)
with open(filename, "w") as f:
for i in range(ND + 1):
p = i / ND
f.write(f"{p} {mc_average(p, num_samples,L)}\n")
def main():
random.seed(0)
if len(sys.argv) != 2:
print("usage: python3 percolation.py systemsize")
return
L = int(sys.argv[1])
num_samples = 1000
mc_all(L, num_samples)
実行してみましょう。
$ python3 percolation.py
usage: python3 percolation.py systemsize
$ python3 percolation.py 8
L08.dat
$ python3 percolation.py 16
L16.dat
$ python3 percolation.py 32
L32.dat
プロットするとこんな感じです。

サイズが大きくなるにつれて、パーコレーション確率の変化が急峻になっていることがわかります。これが有限サイズ効果です。
あとはシステムサイズやサンプル数、何点観測するかを、例えばYAMLやTOMLから指定できるようにすると良いでしょう。
下から書くまとめ
結局、我々は
mainfindunionmc_onestepmc_averagemc_all
という順番で関数を作成して行きました。実際に呼ばれる順番は、
mainmc_allmc_averagemc_onestepunionfind
となるので、概ね呼び出し順と逆向きに開発が進んだことがわかるでしょう。これが「下から」書く例になります。
上から書く例「LAMMPSの出力解析」
LAMMPSという分子動力学法パッケージがあります。初期条件を用意し、LAMMPSに渡して時間発展させ、原子の軌跡(トラジェクトリ)をダンプさせて、そのファイルを解析する、というのがLAMMPSを使った研究のステップになります。
LAMMPSのdumpコマンドにより、トラジェクトリがテキストファイルとして出力されるため、原子の密度や温度といった情報がほしければ、そのファイルを解析する必要があります。以下では、LAMMPSが出力したファイルの解析スクリプトを「上から」書いてみましょう。
LAMMPSの実行
まずはLAMMPSに食わせて実行するためのインプットファイルを作りましょう。適当で良いのですが、とりあえずLennard-Jones原子を、xyz軸にそれぞれ10サイト面心立方格子(FCC)上に配置しましょう。格子定数は2としておきます。FCCは、単位胞あたり4原子あるため、数密度は0.5になります。
その状態でランダムに初速を与え、エネルギー一定条件で1000ステップ実行し、100ステップに一度、温度などの情報を出力し、かつトラジェクトリファイルをダンプするようにしましょう。
units lj
atom_style atomic
boundary p p p
timestep 0.01
# 初期条件の作成
lattice fcc 2.0
region simbox block 0 10 0 10 0 10
create_box 1 simbox
create_atoms 1 box
mass 1 1.0
velocity all create 1.0 1
# 相互作用の指定
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
fix 1 all nve
dump id all custom 100 sample.lammpstrj id type x y z vx vy vz
thermo 100
run 1000
ここではdumpコマンドのデフォルトではなく、ユーザカスタムで「id type x y z vx vy vz」を出力しています。これはID、原子タイプ、そして位置と速度を出力せよ、という命令です。さらに、ファイル名をsample.lammpstrjとしています。拡張子をlammpstrjにすると、VMDで読み込むときに自動判別してくれるので便利です。
これをsample.inputという名前で保存し、LAMMPSに食わせましょう。
$ lmp_serial < sample.input
LAMMPS (20 Nov 2019)
(snip)
0 1 40.272265 0 41.77189 551.3035
100 0.48776863 41.289318 0 42.020788 558.96007
200 0.51621724 41.247985 0 42.022117 558.66534
300 0.49447514 41.288213 0 42.029741 558.96565
400 0.51467079 41.257045 0 42.028858 558.74143
500 0.50585798 41.277221 0 42.035818 558.89749
600 0.51121527 41.268317 0 42.034948 558.83091
700 0.50359229 41.282418 0 42.037617 558.93581
800 0.50639424 41.281686 0 42.041087 558.93668
900 0.51103763 41.275011 0 42.041375 558.886
1000 0.51203328 41.274072 0 42.04193 558.8798
実行した結果、100ステップごとの物理量と、sample.lammpstrjというトラジェクトリファイルが保存されます。このトラジェクトリファイルから原子の情報を抜き出し、例えば温度などの情報を得るのが目的です。
main関数の作成
まずは何も考えずにmain関数を作りましょう。
def main():
pass
if __name__ == "__main__":
main()
ここから開発をスタートします。
read_file関数の作成
トラジェクトリファイルを読み込む関数read_fileを作ります。まずはファイル名を受け取り、それをそのまま出力するだけです。
def read_file(filename):
with open(filename) as f:
for line in f:
print(line, end="")
mainから呼び出します。
def main():
filename = "sample.lammpstrj"
read_file(filename)
この時、ファイル名は必ず変数を経由して渡す癖をつけておくと良いでしょう。
readfile("sample.lammpstrj")
のように直接渡していると、後で修正し忘れることが多いです。
実行しましょう。
$ python3 lammps.py
ITEM: TIMESTEP
0
ITEM: NUMBER OF ATOMS
4000
ITEM: BOX BOUNDS pp pp pp
0.0000000000000000e+00 1.2599210498948732e+01
(snip)
3998 1 11.9697 11.9659 11.3369 0.417186 0.0882677 -0.136933
3999 1 11.9624 11.348 11.9532 -0.315662 0.740397 -1.29379
4000 1 11.3322 11.9701 11.9666 0.377202 -0.691079 0.44533
ちゃんとファイルが読み込めていることがわかります。
さて、LAMMPSのトラジェクトリファイルは
ITEM: アイテム名
内容
ITEM: アイテム名
内容
...
という形式になっています。一番大きな構造はITEM: TIMESTEPで、その中に他にもいろんなITEMが入っています。
- ITEM: TIMESTEP
- ITEM: NUMBER OF ATOMS
- 原子数
- ITEM: BOX BOUNDS pp pp pp
- システムサイズや周期境界条件
- ITEM: ATOMS id type x y z vx vy vz
- 原子のID、位置や速度。これが欲しい情報。
- ITEM: NUMBER OF ATOMS
さて、ファイルを読み進めていって、ITEM: ATOMSという文字列を見つけたら原子の情報を読むようにしなければなりません。まずはITEM: ATOMSの数を数えてみましょう。
def read_file(filename):
with open(filename) as f:
index = 0
for line in f:
if "ITEM: ATOMS" in line:
index += 1
print(index)
実行してみます。
$ python3 lammps.py
11
1000ステップのうち、100ステップごとにダンプを出力しますが、最初にも出力するため、植木算で11フレーム出力されています。正しく取得できているようです。
read_atoms関数の作成
さて、ITEM: ATOMSのあとには、4000行の原子情報が含まれています。それを読み込む関数read_atomsを作りましょう。
まず、read_fileでITEM: ATOMSを見つけたらread_atomsに処理を移します。
def read_file(filename):
with open(filename) as f:
for line in f:
if "ITEM: ATOMS" in line:
read_atoms(f)
次に、read_atomsの中身を作ります。ファイルの構造から、ITEM: TIMESTEPが来るまで読み込めば良いので、とりあえずそこまで表示してみます。
def read_atoms(f):
for line in f:
if "ITEM: TIMESTEP" in line:
exit()
print(line, end="")
ここで、exit()を使って処理を打ち切っています。コードの開発で、「とりあえずここまで実行して確認したい」という時にexit()は便利です。実行してみましょう。
$ python3 lammps.py
1 1 0 0 0 -1.73309 -1.26297 0.876946
2 1 0.629961 0.629961 0 -0.148618 0.123159 -0.977004
3 1 0.629961 0 0.629961 -1.57059 0.627881 0.613321
(snip)
3998 1 11.9692 11.9692 11.3393 -0.444801 0.39035 -1.4034
3999 1 11.9692 11.3393 11.9692 0.50775 0.850047 0.0257805
4000 1 11.3393 11.9692 11.9692 0.218818 -1.37778 -1.041
ちゃんと1フレームの中の原子の情報だけが渡されています。
次に、この行をsplitで分離し、欲しい情報を手に入れます。まずは単純にsplitして、その情報を表示しましょう。
def read_atoms(f):
for line in f:
if "ITEM: TIMESTEP" in line:
exit()
a = line.split()
print(a)
実行します。
$ python3 lammps.py
['1', '1', '0', '0', '0', '-1.73309', '-1.26297', '0.876946']
['2', '1', '0.629961', '0.629961', '0', '-0.148618', '0.123159', '-0.977004']
['3', '1', '0.629961', '0', '0.629961', '-1.57059', '0.627881', '0.613321']
(snip)
['3998', '1', '11.9692', '11.9692', '11.3393', '-0.444801', '0.39035', '-1.4034']
['3999', '1', '11.9692', '11.3393', '11.9692', '0.50775', '0.850047', '0.0257805']
['4000', '1', '11.3393', '11.9692', '11.9692', '0.218818', '-1.37778', '-1.041']
ちゃんと分離できてそうですね。我々が欲しい情報は座標と速度なので、それぞれ取得しましょう。辞書とかを使っても良いですが、ここではAtomクラスを作って、その配列を返すようにしましょうか。
class Atom:
def __init__(self, x, y, z, vx, vy, vz):
self.x = x
self.y = y
self.z = z
self.vx = vx
self.vy = vx
self.vz = vx
def read_atoms(f):
atoms = []
for line in f:
if "ITEM: TIMESTEP" in line:
exit()
a = line.split()
x = float(a[2])
y = float(a[3])
z = float(a[4])
vx = float(a[5])
vy = float(a[6])
vz = float(a[7])
atom = Atom(x, y, z, vx, vy, vz)
atoms.append(atom)
print(atom)
例によって、わざと間違えています。
ついでにちゃんとAtomオブジェクトができたか、printで確認しています。実行してみましょう。
$ python3 lammps.py
(snip)
<__main__.Atom object at 0x7f85efd6b520>
<__main__.Atom object at 0x7f85efd6b580>
<__main__.Atom object at 0x7f85efd6b5e0>
<__main__.Atom object at 0x7f85efd6b640>
<__main__.Atom object at 0x7f85efd6b6a0>
<__main__.Atom object at 0x7f85efd6b700>
クラスを表示するとこんな感じになってしまい、中身がわかりません。そこで、Atomクラスに__str__メソッドを追加しておきましょう。
class Atom:
def __init__(self, x, y, z, vx, vy, vz):
self.x = x
self.y = y
self.z = z
self.vx = vx
self.vy = vx
self.vz = vx
def __str__(self):
return f"{self.x} {self.y} {self.z} {self.vx} {self.vy} {self.vz} "
すると、実行結果はこんな感じになります。
0.0 0.0 0.0 -1.73309 -1.73309 -1.73309
0.629961 0.629961 0.0 -0.148618 -0.148618 -0.148618
0.629961 0.0 0.629961 -1.57059 -1.57059 -1.57059
0.0 0.629961 0.629961 1.49597 1.49597 1.49597
(snip)
11.9692 11.9692 11.3393 -0.444801 -0.444801 -0.444801
11.9692 11.3393 11.9692 0.50775 0.50775 0.50775
11.3393 11.9692 11.9692 0.218818 0.218818 0.218818
これと先ほどの生の出力、
$ python3 lammps.py
1 1 0 0 0 -1.73309 -1.26297 0.876946
2 1 0.629961 0.629961 0 -0.148618 0.123159 -0.977004
3 1 0.629961 0 0.629961 -1.57059 0.627881 0.613321
(snip)
3998 1 11.9692 11.9692 11.3393 -0.444801 0.39035 -1.4034
3999 1 11.9692 11.3393 11.9692 0.50775 0.850047 0.0257805
4000 1 11.3393 11.9692 11.9692 0.218818 -1.37778 -1.041
を比較し、おかしいことに気づきます。自分が取得したデータは、速度成分がなぜかすべて一致しています。ここで初めて以下の間違いに気づきます。
class Atom:
def __init__(self, x, y, z, vx, vy, vz):
self.x = x
self.y = y
self.z = z
self.vx = vx
self.vy = vx # コピペミス
self.vz = vx # コピペミス
直しておきましょう。
class Atom:
def __init__(self, x, y, z, vx, vy, vz):
self.x = x
self.y = y
self.z = z
self.vx = vx
self.vy = vy
self.vz = vz
def __str__(self):
return f"{self.x} {self.y} {self.z} {self.vx} {self.vy} {self.vz} "
実行します。
$ python3 lammps.py
0.0 0.0 0.0 -1.73309 -1.26297 0.876946
0.629961 0.629961 0.0 -0.148618 0.123159 -0.977004
0.629961 0.0 0.629961 -1.57059 0.627881 0.613321
(snip)
11.9692 11.9692 11.3393 -0.444801 0.39035 -1.4034
11.9692 11.3393 11.9692 0.50775 0.850047 0.0257805
11.3393 11.9692 11.9692 0.218818 -1.37778 -1.041
生データと比べて、正しく値が取得できたことがわかります。
では、作成されたatoms配列を返すように修正しましょう。
def read_atoms(f):
atoms = []
for line in f:
if "ITEM: TIMESTEP" in line:
return atoms
a = line.split()
x = float(a[2])
y = float(a[3])
z = float(a[4])
vx = float(a[5])
vy = float(a[6])
vz = float(a[7])
atom = Atom(x, y, z, vx, vy, vz)
atoms.append(atom)
実はこのコードにはまだ間違いがありますが、わかりますか?
とりあえず間違いには気づかなかったとして、read_atomsを呼んでいるread_filesで、atomsの配列を作ることにしましょう。
def read_file(filename):
frames = []
with open(filename) as f:
for line in f:
if "ITEM: ATOMS" in line:
atoms = read_atoms(f)
frames.append(atoms)
return frames
フレームごとにatomsという配列が渡されるので、その度にframesという配列に追加しています。今回は4000原子なので、各フレームごとにatomsの長さは4000になるはずです。表示してみましょう。以下のようなmain関数を書きます。
def main():
filename = "sample.lammpstrj"
frames = read_file(filename)
for atoms in frames:
print(len(atoms))
実行してみましょう。
$ python3 lammps.py
4000
4000
4000
4000
4000
4000
4000
4000
4000
4000
Traceback (most recent call last):
File "lammps.py", line 48, in <module>
main()
File "lammps.py", line 44, in main
print(len(atoms))
TypeError: object of type 'NoneType' has no len()
最後10フレームは正しく取得できていますが、最後のフレームが変です。これは、read_atomsのフレーム終了判定が、「次のITEMS: TIMESTEPを見つけた時」になっているためです。最後のフレームは、ITEMS: TIMESTEPが来る前にファイルが終わってしまいます。したがって、read_atomsを以下のように書き換えないいけません。
def read_atoms(f):
atoms = []
for line in f:
if "ITEM: TIMESTEP" in line:
return atoms
a = line.split()
x = float(a[2])
y = float(a[3])
z = float(a[4])
vx = float(a[5])
vy = float(a[6])
vz = float(a[7])
atom = Atom(x, y, z, vx, vy, vz)
atoms.append(atom)
return atoms # ここを追加
もう一度実行してみましょう。
$ python3 lammps.py
4000
4000
4000
4000
4000
4000
4000
4000
4000
4000
4000
正しく取れたようですね。
temperature関数の実装
原子の情報すべてが取れたので、各フレームごとの物理量を計算してみましょう。ここでは、一番計算が簡単な温度を計算してみます。atoms配列を受け取り、温度を返す関数temperatureを作ります。運動エネルギーを
def temperature(atoms):
N = len(atoms)
K = 0.0
for a in atoms:
K += a.vx**2
K += a.vy**2
K += a.vz**2
T = K / 3.0 / N
return T
表示してみましょう。
def main():
filename = "sample.lammpstrj"
frames = read_file(filename)
for atoms in frames:
print(temperature(atoms))
実行してみます。
$ python3 lammps.py
0.9997499474435989
0.4876466734158087
0.5160881727924679
0.4943515587457448
0.5145421433156472
0.50573154319418
0.5110874926599625
0.5034663387203961
0.5062676018157087
0.5109098208368243
0.5119052941726792
LAMMPSが吐いてきた温度と比べます。
$ lmp_serial < sample.input
LAMMPS (20 Nov 2019)
(snip)
0 1 40.272265 0 41.77189 551.3035
100 0.48776863 41.289318 0 42.020788 558.96007
200 0.51621724 41.247985 0 42.022117 558.66534
300 0.49447514 41.288213 0 42.029741 558.96565
400 0.51467079 41.257045 0 42.028858 558.74143
500 0.50585798 41.277221 0 42.035818 558.89749
600 0.51121527 41.268317 0 42.034948 558.83091
700 0.50359229 41.282418 0 42.037617 558.93581
800 0.50639424 41.281686 0 42.041087 558.93668
900 0.51103763 41.275011 0 42.041375 558.886
1000 0.51203328 41.274072 0 42.04193 558.8798
この2列目が温度です。ほぼ一致しており、正しく温度が計算できたことがわかります。
上から書くまとめ
我々は
mainread_fileread_atoms
という順番で関数を作成しました。これは呼び出し順と一致しており、「上から書いた」ことに対応しています。
まとめ
コードを書く時には、小さいパーツをまず書いてから組み上げていく「下から」書くパターンと、大きな枠組みを先に作って中を埋めていく「上から」書くパターンがあります。どちらで書くかは場合によりますし、上下両方から組み進める場合もあるでしょう。
いずれにせよ、少し書くたびに、しつこいくらいに中身をprintで表示し、想定どおりにプログラムが組めているかどうか確認するのが重要です。「これは大丈夫だろう」を放置しておくと、後で数倍、数十倍のロスとなって自分に襲いかかって来ます。自分は「さすがに大丈夫のはず」というコードをテストするようになってから、ようやく「プログラムが普通に組めるようになったな」と感じました。
この小文が「プログラムはなんとなくわかったけど、ゼロから組むのにどうすればよいかわからない」という初心者の助けになれば幸いです。
Discussion