分子動力学法でなるべく精密に指定の密度の初期条件を作る方法
概要
分子動力学法で、原子を指定密度で初期配置したいことがよくあります。適当に配置してしまうとエネルギーが爆発してしまったりするので、なるべく原子同士は離して配置したいですね。ここでは、そんな配置手段の一つとして、格子欠陥のある面心立方格子に組む方法を紹介します。
欠陥のない面心立方格子
多くの単純原子が面心立方格子(Face Centered Cubic, FCC)状の固体を持ちます。特に、ネオン、アルゴン、クリプトンといった希ガスの固体がFCCとなります。古典分子動力学法で広く使われているLennard-Jones相互作用は希ガスの分子間力を模したものなので、やはり固体はFCCとなります。まずはなるべく指定の密度に近いFCCを組むコードを組んでみましょう。
簡単のため、シミュレーションボックスは一辺
となります。格子欠陥がない場合、指定の密度に最も近い
これは先ほどの式を
です。
import numpy as np
def get_lattice_number(L, rho):
m = np.floor((L**3 * rho / 4.0)**(1.0 / 3.0))
drho1 = np.abs(4.0 * m**3 / L**3 - rho)
drho2 = np.abs(4.0 * (m + 1)**3 / L**3 - rho)
if drho1 < drho2:
return m
else:
return m + 1
この方法だと、
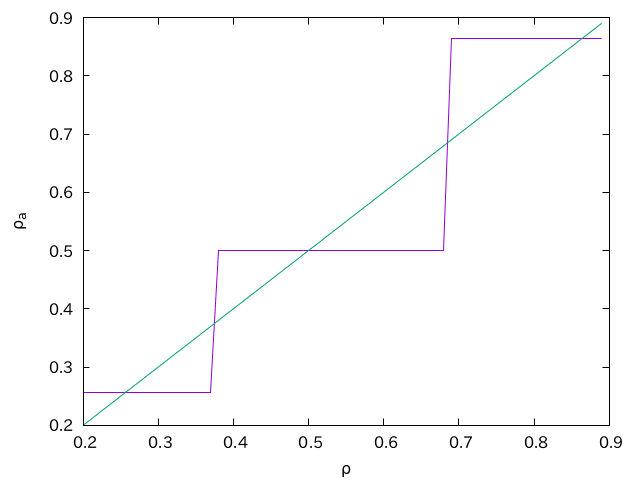
これをなんとかしましょう、というのが本稿の主題ですが、それはそれとして指定の格子数、格子定数でFCCに組むコードを作っておきましょう。格子定数
def make_fcc_pure(L, rho):
m = get_lattice_number(L, rho)
a = L / m
ha = a * 0.5
atoms = []
for i in range(m**3):
ix = i % m
iy = (i // m) % m
iz = i // (m * m)
x = ix * a
y = iy * a
z = iz * a
atoms.append((x, y, z))
atoms.append((x + ha, y + ha, z))
atoms.append((x + ha, y, z + ha))
atoms.append((x, y + ha, z + ha))
return atoms
素直に三重ループ回しても良いですが、あまりインデントが深くなるのもアレなのでシングルループに展開しました。
atoms = make_fcc_pure(10.0, 0.5)
for a in atoms:
print(f"{a[0]} {a[1]} {a[2]}")
実行結果はこんな感じです。
0.0 0.0 0.0
1.0 1.0 0.0
1.0 0.0 1.0
0.0 1.0 1.0
(snip)
8.0 8.0 8.0
9.0 9.0 8.0
9.0 8.0 9.0
8.0 9.0 9.0
なんかできてそうですね。
欠陥のある面心立方格子
さて、欠陥のない面心立方格子では、
Pythonであればrandom.sampleを使うことで指定のリストから指定の数だけ重複無しにランダムに要素を取り出したリストを作ることができます。素直に実装するとこんな感じになるでしょう。
import numpy as np
import random
def get_lattice_number(L, rho):
m = np.ceil((L**3 * rho / 4.0)**(1.0 / 3.0))
return int(m)
def make_fcc_pure(L, rho):
m = get_lattice_number(L, rho)
a = L / m
ha = a * 0.5
atoms = []
for i in range(m**3):
ix = i % m
iy = (i // m) % m
iz = i // (m * m)
x = ix * a
y = iy * a
z = iz * a
atoms.append((x, y, z))
atoms.append((x + ha, y + ha, z))
atoms.append((x + ha, y, z + ha))
atoms.append((x, y + ha, z + ha))
return atoms
def make_fcc_defect(L, rho):
atoms = make_fcc_pure(L, rho)
n = int(rho * L**3)
return random.sample(atoms, n)
先ほどと違って、かならず指定密度「以上」の欠陥無し格子を作るために、get_lattice_numberで天井関数を使っています。後は、欠陥無しのFCCの原子リストを作ってから、random.sample(atoms, n)で欲しい数だけ抜き出すだけです。
atoms = make_fcc_defect(10.0, 0.489)
for a in atoms:
print(f"{a[0]} {a[1]} {a[2]}")
実行してみます。
$ python3 defect.py | wc
489 1467 5868
ちゃんと489原子が出力されています。プロットしてみましょう。
まず、欠陥無しの場合(

欠陥ありの場合(

よく見ると、いくつか原子が欠けているのがわかると思います。実際には、欠陥無しの場合に比べて原子が11個欠けており、中途半端な密度
まとめ
分子動力学法において、指定の密度で、なるべく「ちゃんとした」状態の初期条件を簡単に作る方法を紹介しました。初期配置を適当にすると計算が不安定になったり緩和が遅くなったりすることがあります。とりあえずFCCに組んで粒子を適宜間引く方法は楽で汎用的なので、覚えておくとたまに便利かもしれません。
Discussion