ASEライブラリを使ってグラフェン分子を書く
はじめに
この記事ではPython上でグラフェンシートを描いていきます。
化学で用いられるLAMMPS[1]などのシミュレーションでは系の大きさ、存在する分子(あとは各種シミュレーション条件)などを指示するために専用のファイルを作る必要があります。
多くは論文のリファレンスなどでシミュレーションに関する詳細が載っているためにご自身のパソコン上でそれらのシミュレーションを再現する際にはあまり何も考えず、コードを確認し、必要ならばチューニングを行い、それをコピペすれば動くでしょう。また単に分子がほしい際にはGaussian[2]上で分子を書けばいいですし、なんなら構造最適化までできますよね...
しかし、この記事ではそういったコピペに頼らず、構造を書いてみようよ!といったものです。つまり 興味だけでグラフェンシートを描こうよ! といったものです。
さて。LAMMPSなどのシミュレーションソフトを念頭においたとき、その入力形式に合わせたファイルを手軽に出力するための強力なツールがPython上に存在します。
それが題名にもある、Atomic Simulation Environment:ASE というライブラリ[3]です。
このモジュールの基本的な使い方は以下のサイトに載っています。
※幅広い読者が読めるように専門用語などには脚注[4]を付けました。参考にしてください。
前提知識の確認
グラフェンの構造
コードを書くにしてもグラフェンの立体構造がわからなければ何もできません。なのでまずはグラフェン graphene の基本的なお話をしましょう。
グラフェンは炭素のみからなるシート状の構造体です。注意してほしいのはグラファイトという物質とは違うということです。グラファイトはこのグラフェンがいくつも重なったものです。[5]
グラフェンが結晶か否か、みたいな話はヤヤコシイので今回は濁しますが、2次元平面で周期的な構造を持っているのは明らかでしょう。グラフェンは周期的なハニカム構造を持っています。

つまり、座標平面上に落としこむ際には二次元あれば大丈夫です。
次に必要なのは炭素同士の結合距離です。ここでは1.42Å として考えましょう。(1Å=10^-10 m) )です。[6]
最後にコードで実装するというからにはいちいち手計算をして座標を入力するわけにはいきません。繰り返しの基本構造を考えましょう。
とはいっても、この分子は対象性が半端ないので結構どんな基本構造でもうまくいってしまいます。
しょうがないので六角形構造の一編、つまり2つの炭素原子のみを基本構造にしたいと思います。
うち一つを原点(0,0,0)におき、もう一つをy軸上に結合距離だけ離した位置(0,1.42,0)に置きます。
これでオッケーです。z軸はシミュレーションが3次元で行う(当たり前)ので書き込んでいますが今回は単に1枚のグラフェンシートを描くことが目的なので常にz=0 です。
ついでに格子定数について考えておきましょう。ここでいう格子定数はある炭素原子に注目したとき、対面に存在する炭素原子までの距離を指しています。これは単にnp.sqrt(3)*1.42 です。
ASEモジュールのコード
ASEモジュールで用いられるコードについて考えていきましょう。
Atomsクラス
AtomsクラスはASEモジュールの心臓部にあたるものです。
シミュレーションにおける系の設定を一挙に設定してしまうものです。
大きく5つの引数を持ちます。
graphene = Atoms(symbols, positions, cell, pbc=[True, True, False])
変数名 graphene にAtoms を入れていますがこのときgraphene に入っているのはシミュレーションの各種条件です。
symbols にはシミュレーションに用いる原子を必要な個数
position にはそれらの原子の3次元座標
cell はシミュレーションセルのことで、つまりシミュレーションを行うときの系の大きさです。周期境界条件を置くことで実際の系のようにすることができます。
pbc は周期境界条件の設定です。今回はグラフェンが2次元なのでx,y は周期境界条件を入れますがzにいはいれません
今回は繰り返しの基本構造がほしいのでユニットセルのみをAtomsで設定します。
余談ですがAtoms はその分子の速度も設定できます。
repeat
repeat は基本構造を拡大するときにつかうメソッドです。上で設定したようなAtoms の条件を欲しいサイズに拡大するときに使います。返り値もAtoms になります。(つまりセルで設定した周期境界条件などを引き継ぎます)
スーパーセル = ユニットセル.repeat((nx, ny, nz))
このメソッドによって上で設定したユニットセルをスーパーセルに拡大することができます。
write
ここはシミュレーションに関係ありませんが、最後できたファイルをwrite で書きだすことができます。今回は.xyz ファイルで書きだしますが、.png などで書きだすこともできるそうです。
write("出力ファイル名", 出力するAtomsクラスの変数))
これで最低限の情報を載せたと思います。では実装へ。
コーディング
はじめに全体像です。全体で43行ほど
from ase import Atoms
from ase.io import write
import numpy as np
import matplotlib.pyplot as plt
# --- パラメータ ---
bond = 1.42 # C-C bond length [Å]
a = np.sqrt(3) * bond # graphene lattice constant
nx, ny = 10, 10 # 繰り返し数
# --- セルベクトル ---
cell = np.array([
[a, 0, 0],
[a/2, a*np.sqrt(3)/2, 0],
[0, 0, 20.0] # 真空を20 Å確保
])
# --- 原子の種類と位置(2原子) ---
positions = [
(0.0, 0.0, 0.0),
(0.0, bond, 0.0)
]
symbols = ["C", "C"]
# --- Atoms オブジェクト作成 ---
graphene = Atoms(symbols=symbols, positions=positions, cell=cell, pbc=[True, True, False])
# ---スーパーセルの設定---
supercell = graphene.repeat((nx, ny, 1))
# --- 書き出し ---
write("graphene_5x5.xyz", supercell)
# --- 可視化(x–y 平面) ---
pos = supercell.get_positions()
plt.figure(figsize=(6,6))
plt.scatter(pos[:,0], pos[:,1], s=20, c="black", alpha=0.7)
plt.gca().set_aspect("equal", "box")
plt.xlabel("x (Å)")
plt.ylabel("y (Å)")
plt.title("Graphene 5x5 supercell (bond=1.42 Å)")
plt.show()
ライブラリの確認
はじめにPython の使用ライブラリを確認します。
from ase import Atoms
from ase.io import write
import numpy as np
import matplotlib.pyplot as plt
上記で述べたシミュレーション設定のAtomsと書き出しのwrite を入れています。
あと一部の計算・行列の入力と最後の描画のためにnumpy,matplotlib.pyplot を入れています。
ase.io のモジュールがファイルの読み込み、出力を行うものです。
ase はAtoms のようなシミュレーションの基本設定を扱うものです。
パラメータの設定
各種定数を定義します。
bond = 1.42 # C-C bond length [Å]
a = np.sqrt(3) * bond # graphene lattice constant
nx, ny = 10, 10 # 繰り返し数
今回は縦横に10ずつユニットセルを並べます。
ユニットセルの定義
ユニットセルの定義、つまり繰り返し構造の基本となるシミュレーションの基本単位の箱を設定します。
cell = np.array([
[a, 0, 0],
[a/2, a*np.sqrt(3)/2, 0],
[0, 0, 20.0] # 真空を20 Å確保
])
3つの基底ベクトルを設定しています。3つ目はz軸方向のものです。
原子の種類と位置
positions = [
(0.0, 0.0, 0.0),
(0.0, bond, 0.0)
]
symbols = ["C", "C"]
ふたつの原子とももちろん炭素!一つは原点、もう一つはy軸上に結合距離分離して置く。
Atoms の設定とスーパーセルの設定
graphene = Atoms(symbols=symbols, positions=positions, cell=cell, pbc=[True, True, False])
supercell = graphene.repeat((nx, ny, 1))
nz の繰り返し単位は1つ。つまり1層だけでいい。ここをいじるとグラファイトが完成します。
書き出し
write("graphene_10x10.xyz", supercell)
ここで出力ファイルの拡張子を.xyz に設定
##図示
pos = supercell.get_positions()
plt.figure(figsize=(6,6))
plt.scatter(pos[:,0], pos[:,1], s=20, c="black", alpha=0.7)
plt.gca().set_aspect("equal", "box")
plt.xlabel("x (Å)")
plt.ylabel("y (Å)")
plt.title("Graphene 10x10 supercell (bond=1.42 Å)")
plt.show()
結果
matplotlib
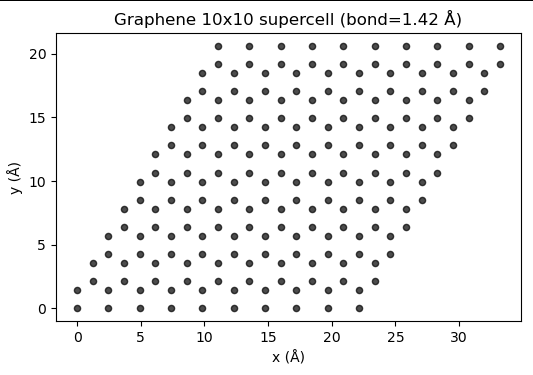
Python上ではとりあえずこのような出力ができました。
VESTA

いいかんじ
VMD
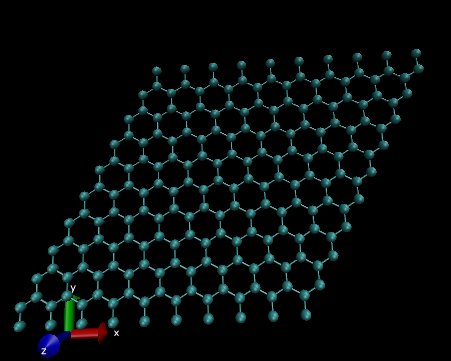
おわりに
graphene を書き、.xyzファイルでの書き出しはできましたよということでした。
ありがとうございました。
-
LAMMPSは化学において各種シュミレーションを行うオープンソースソフトウェア ↩︎
-
Gaussianは第一原理計算を利用した化学のシミュレーションソフトウェア ↩︎
-
ライブラリはPythonにおける便利機能をまとめた入れ物のようなものです。何かコードを書く際に書きたいものによって最初に導入するライブラリをいろいろと考える必要があります ↩︎
-
脚注footnoteとは本文下部に設ける注のことである ↩︎
-
今回は簡単のためグラフェンを書きます ↩︎
-
Å(読み:オングストローム)は原子や分子の大きさを扱うときに用いられる長さの単位です。非SI単位系ですが頻繁に使われています。SI単位系的には接頭辞のnをつけて0.1nmに当たります。 ↩︎
Discussion