【論文紹介】PharmAgents解説
タイトル:PharmAgents: Building a Virtual Pharma with Large Language Model Agents
※ 本ページの図は特筆がない限り全て本論文から引用しています。
今回は、「PharmAgents: Building a Virtual Pharma with Large Language Model Agents」という論文を紹介したいと思います。
概要
PharmAgentsはLLMベースのマルチエージェントを活用した製薬バーチャルエコシステムです。製薬におけるDrug discoveryを対象とし、そのワークフローをターゲット同定から前臨床的評価までLLMベースエージェントがシミュレーションします。一気通貫でDrug discoveryのプロセスをLLMでシミュレーションする初の取り組みになります。特に、各ステップにおいて説明可能AIとも通ずる解釈性が向上することが期待されています。それだけでなく、ユーザとのインタラクションやLLMの自己学習、あるいは薬のライフサイクルマネジメントなどを包括的に扱えるシステムを実現するポテンシャルを秘めている取り組みと言えます。
要するに、通常5年以上を要する臨床試験までのプロセスを「Virtual Pharma」と名づけられたこのシステムで高速に実現しようという試みです。

AI エージェントの設計方針について
近年AIエージェントの研究ではwell-definedな役割を定め、それぞれの役割を連携させて最終成果物を生成する取り組みをよく見かけます。例えばコード生成の例では、PM・開発者・品質担保エンジニア…といった具合です。それと同様に、PharmAgentsにおいても、Drug discoveryのプロセスを
① target discovery(ターゲット同定)
② lead identification(リード化合物の同定)
③ lead optimization(リード最適化)
④ preclinical evaluation(前臨床的評価)
という4つのステージに整理し、それぞれを専門のエージェントが担います。
各ステップは、製薬業界の用語です。背景知識として、これらを整理しておきましょう。詳細については専門家による記事などを参考にしてください。
- ターゲット:病気の進展や症状を抑えることができうる分子のこと
- ヒット化合物:スクリーニングによって得られる候補となる化合物
- リード化合物:ヒット化合物の中でも更に活性が確認された良さそうな候補となる化合物
- リード最適化:例えば官能基修飾などによりリード化合物を安全性等の観点から改善したもの
- 前臨床試験(非臨床試験):治験などの臨床試験の前に行われる研究段階。典型的には動物実験。
Virtual Pharmaフレームワークのデザイン
いよいよ本丸のエージェントシステムがどう作成されているかについて見ていきます。Figure2によると、前述のように4つの段階からなるワークフローとして構成されていますね。
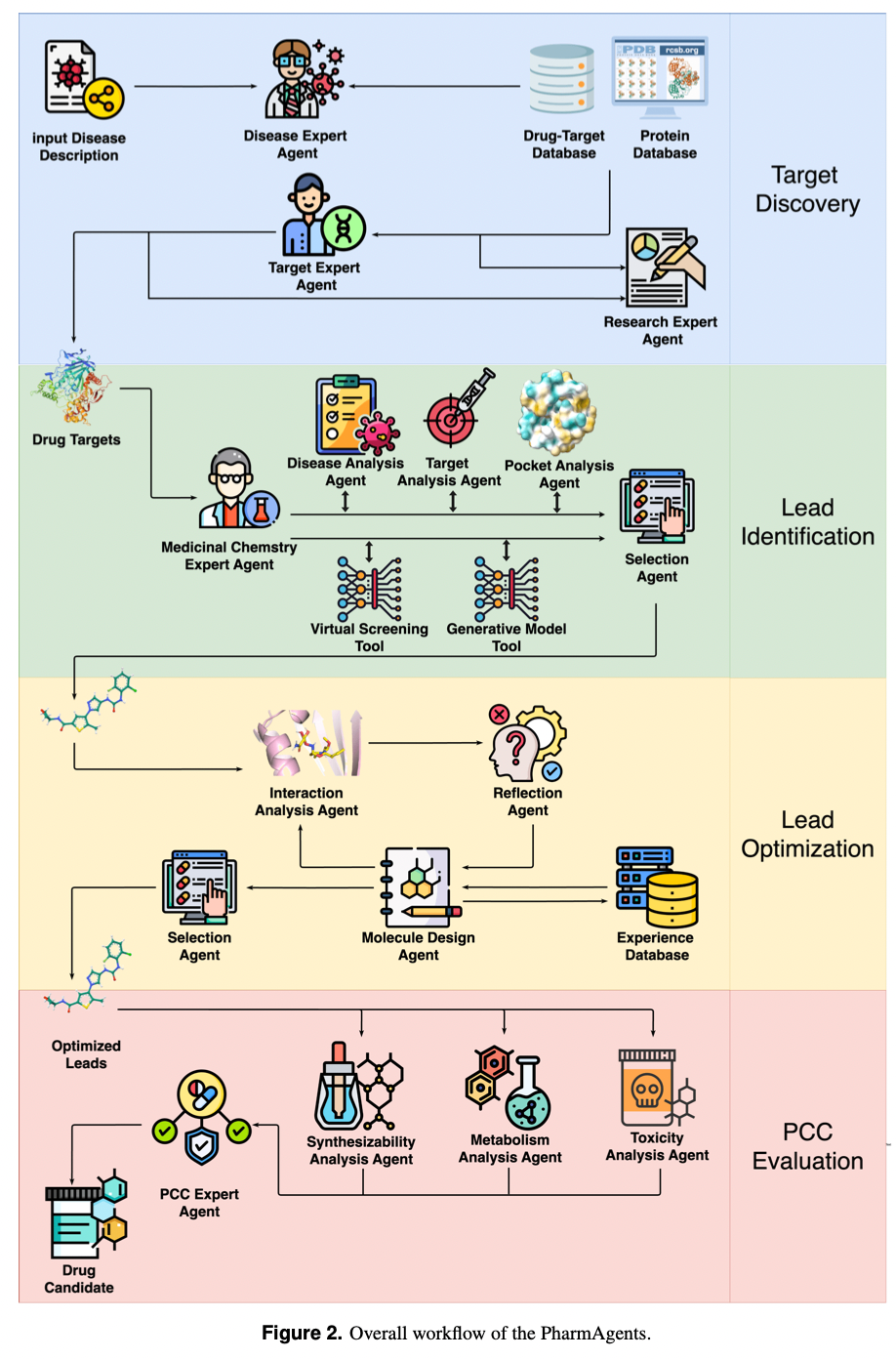
Phase 1:Target Discovery(創薬ターゲット探索)
最初はTarget Discoveryのフェーズですね。このフェーズの中でも2段階に分割されています。まず、入力としては病名を想定しているようです。

Disease expert agentと名付けられたAgentがDrug Target DatabaseとUnified Protein Databaseにアクセスしながら、類似の病気とそれに対応するターゲットを調べ上げます(この論文では3種類)。続いてStructure Expert Agentと名付けられたAgentが関連文献やリガンド情報に基づきPDB IDと呼ばれるIDをリストアップします。PDBというのはRCSB Protein Data Bankというタンパク質に関する既存のデータベースのことです。このデータは抽出されたタンパク質について、簡単な説明文や関連論文のアブスト、共結晶リガンドの名称などが含まれます。
**「リガンド(ligand)」って何?**
リガンドとは、タンパク質のある特定の部分(活性部位など)に結合する分子です。
薬の有効成分も、よくリガンドになります。
**「共結晶(co-crystal)」って何?**
「タンパク質と小さな分子(リガンド)が一緒に結晶化した構造」のことです。
この構造は、X線結晶構造解析などで決定され、タンパク質がどのようにリガンドと結合しているかを3Dで見ることができます。
**「共結晶リガンド」とは?**
たとえば:
「この酵素はどんな薬とくっつくか?」を調べるために、
薬の候補(=リガンド)と酵素を一緒にして結晶にして、
X線解析で、どこにどう結合するかを確認します。
このときに結合していた薬分子(リガンド)が「共結晶リガンド」です。
特にこの共結晶リガンドの名前はこの後のステップにおける選択基準として大変に重要な役割を通常果たします。さらにここからPDB IDで示された候補群に対し以下のフィルタリングを行います。このフィルタリングアルゴリズムは基本的にはStructureExpertがターゲットへの結合性をもとに判断し、Top10を決定します。ただ一度それを行うだけでは心もとないので、まずPDB候補を100個ずつのグループに分け、① 各グループのTop10を選出、② 各グループ内で逆順に並べ替えた上で同様にTop10を選出し、①と②で重複したPDBを残すという小さな工夫が見られます。最終的に各グループの選ばれしPDB候補等を全て集め、もう一度同様に選別をします。網羅性の向上や不確実性の抑制のためにこのような手順を取り入れたものだと考えられます。加えて、Unified Protain Databaseを参照しながら偏りを防いでいます。また下図には含まれていませんが、後で登場するResearch Expertというエージェントがいて、このプロセスでの履歴は全てこのResearch Expertに報告がされることになっています。

Phase 2:Lead Identification(リード化合物の特定)
このプロセスではあらゆるツールを活用してターゲットと反応しうる分子を見出します。活用するツールの1つはDecompDiffというdrug design modelの1つです。もう1つはDrugCLIPという創薬特化CLIPモデルです。
ここで簡単に先行研究で作成されたこれらのモデルがどういうものなのか確認しておきましょう。
DecompDiff: Diffusion Models with Decomposed Priors for Structure-Based Drug Design とは
Strucure-Based Drug Designという分子立体構造を基に化合物を設計する創薬手法があります。その中で2024年に登場し、CrossDocked2020というデータセットによるベンチマーク評価でSOTAを達成した拡散モデルがDecompDiffです。Diffusionモデルは画像生成で一躍知名度を上げましたが、3D分子構造生成の分野では原子の位置座標と種類を生成するのに使用されてきました。一方で原子間の化学結合については事後的なアルゴリズムにより決定されることが多かったようです。それに対しDecompDiffではBond Diffusionというものを導入し、分子中の「原子 = ノード」と「結合 = エッジ」の双方をDiffusionで生成するアプローチを新たに提案しています。
DrugCLIP:Contrastive Protein-Molecule Representation Learning for Virtual Screening とは
Virtual screeningという創薬ターゲットと化合物間の相互作用を数値シミュレーションするプロセスで利用可能なドメイン特化型CLIPモデルです。CLIPというのはOpenAIを中心にtext-to-imageなどのパーツとして一躍有名になったマルチモーダルモデルです。異なるドメインのペア(画像とテキストなど)を対応づけることを目的とし、Contrastive Learningを用いて学習されます。Contrastive learningとは「近いものは近づけ、遠いものは遠ざける」という意図で行われる学習のことです。

DrugCLIP (Gao et al., 2023, NeurIPS) Figure 1
このような既存モデルの活用だけではなく、さらにLLM-guided molecule generationと称し、Disease Analysis agent・Target Analysis Agent・Pocket Analysis Agentや、実際の化学者が蓄積された知識や症状/ターゲット/分子間反応への深い理解をもとに薬を設計していく思考過程を模したLLMシミュレーションも追加されています。
以上をまとめると創薬手法の3つのアプローチ**「構造ベース・バーチャルスクリーニング・LLM生成」**を織り混ぜ、最後にさらにまたLLMを用いて有望な分子構造を選別するのがLead Identificationフェーズの全貌です。分子構造をLLMで扱う際は下図左下部のプロンプトに記載されているようにSMILES記法(化学構造をASCII符号の英数字で文字列化した汎用的な化合物情報フォーマット)が利用されています。
Phase 3:Lead Optimization(リード最適化)
リード最適化は候補として有望な分子を"viable drug candidate" = 実現可能な薬の候補へと変換するプロセスのことです。
- ↑増強したい: 結合親和性, ドラッグライクネス(医薬品らしさ), 合成の実現性, 構造の完全性
- ↓抑制したい:オフターゲット効果(本来の標的分子以外と相互作用してしまうこと)、毒性
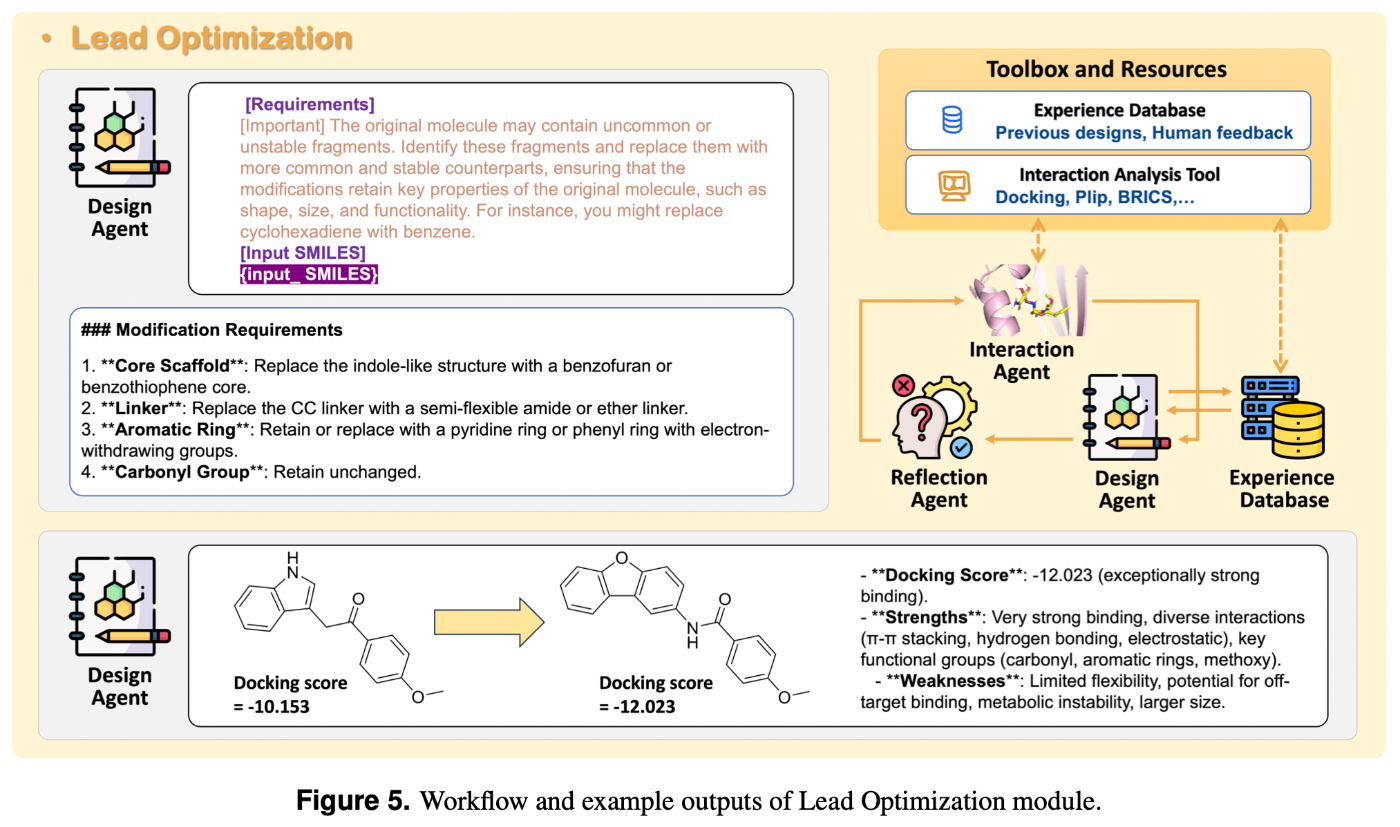
従来はシステマティックに分子構造 を変更してきましたが, VirtualPharmaではエージェントたちが繰り返し協議し分子構造を改善していきます。
を変更してきましたが, VirtualPharmaではエージェントたちが繰り返し協議し分子構造を改善していきます。
登場人物は以下の5人です。
- Interaction analysis agent
- Design agent
- Generation agent
- Reflection agent
- Selection agent
まずInteraction analysis agentがdocking softwareとPLIPを利用して対象の分子とタンパク質ポケットの結合や相互作用を調べます。次にDesign agentが改善方法を提案し、Generation agentが次の候補分子を生成します。その後, Interaction analysis agentが再び登場します。改善後の分子の結合を調べ、Reflection agentがその改善によって満たしてほしい性質が満たされたかを評価します。
この一連のプロセスが大体5回ほど繰り返され、分子構造が改善されることを期待します。
最終的に、Selection agentが生成された候補分子全てを分子間相互作用や薬関連の性質の面から判断し、ベストのものを選び出します。
Phase 4:Preclinical candidate evaluation(前臨床的評価)
実際の薬の開発では、薬の効能が潜在リスクを上回っていることが肝要です。最後のプロセスとして毒性と合成可能性の側面から編み出された候補分子のことを評価します。ここでは2つのエージェントによる評価がパラレルに実施されます。
まず、Metabolism and Toxicity Assessment AgentはMetaTransという高性能なモデルを用いて毒性を評価します。内部的にはTOXRICという10000以上の分子のLD50(半数致死量)を記録したデータベースを活用しますが、これはより良いデータベースに差し替えて性能向上を図っても良い部分です。未知の化合物の毒性を予測する際は典型的には類似する5つの化合物を参考にします。化合物間の類似度は「タニモト係数」などで測ることができるそうです。
もう一つのエージェントがSynthesis Assessment Agentです。こちらのエージェントは候補化合物の合成可能性を評価します。化学物質の合成ルートを自動で提案する逆合成解析という領域です。内部的にはUAlignというディープラーニングモデルが利用されています。
それだけではなくUAlignが弾き出した合成ルートはLLMによって評価され、どれくらい合成可能であるかスコアが0~100で算出されます。この工夫はLLMが内在的に有する逆合成解析に関する知識を利用できることを期待しています。

上図には明示されていませんが最終的にはReport Assessment Agentという別のエージェントに各エージェントの結論が全て統合的に入力された上で、臨床開発に回すに値するか否かを二値分類で判定するようです。例えば薬としては高性能で有害性も低い候補分子であっても、もし合成可能性が高すぎれば、このエージェントによって弾かれる可能性があります。
以上で病名(input_disease)を入力し、候補分子を抽出した上で合格か不合格かを二値分類結果を出力するまでの一連のプロセスが完成しました。
結果
気になるのは以上のような壮大なフレームワークが実際にワークするのかという点です。本論文では各Phaseに対して結果が示されています。
Target discovery
GPT-4o, GPT-4o-mini, DeepSeek-V3, DeepSeek-R1の4種類のLLMに対し、9種類の疾病を入力した結果、微妙な違いはあるものの概ね似たような化合物をターゲット候補として出力したようです。定量的な評価は難しいですが、Human expertによる評価でも合理的な結果になりました。特に「Drug Target Database」にアクセスできることが良い判断につながりました。
アトピー性皮膚炎を例に入力した際の結果は以下でした。これら18個の結果のうち、16個は生物学的に妥当な結果とのことです。

Lead Identification & Lead Optimization
定量評価の詳細は割愛しますが、DecompDiffやMolCraftなどのstructure-based drug design modelのSOTA(state-of-the-art)モデルに対し、成功率で3倍近くの改善を達成しました。これらの評価は主にGPT-4oを用いて実行されましたが、GPT-4o-miniやDeepSeek-V3, DeepSeek-R1も試されています。結果的には、GPT-4o-miniはdocking scoreで劣りました。また、DeepSeek-V3は初期の候補物質からの変更が少なく抑えられる傾向がある一方で、reasoningモデルであるDeepSeek-R1はより広範な分子候補を探索し変更幅が大きくなる傾向がありました。
PCC Evaluation
まず毒性予測に関してですが、結果的にはGPT-4oが最も適したモデルとなりました。高い予測率や低い過小評価率を示しました。一方、合成可能性に関する評価については、LLMが算出したconfidence score(0〜100)とSAスコア(Synthetic Accessibility score の略, 1~10)とのピアソン相関係数を主に調べています。結果的には少し信頼できない部分もあるものの, GPT-4o-miniが-0.645という最も強い負の相関を示しました。既存のSAスコア自身が完璧な指標では勿論ないので、解釈が難しい部分となります。
まとめ
今回は、マルチエージェントによる新薬開発のパイプラインであるPharmAgentsの枠組みをまとめました。LLMを用いたエージェントをシームレスに繋ぎ段階的にタスクをこなさせるメリットの一つは(統計的な妥当性などはともかくとして)言語出力を追えるおかげでユーザである人間にとって解釈性や透明性が高く保たれる点にあります。特にリード化合物の特定やリード最適化においては、既存のSOTA手法を大きく改善することができました。このようなシステムは最近話題にとしてよく聞きますが、「Self-evolution」のように過去から学び続けどんどん賢くなっていく、どんどん進化していくという期待も存在します。PharamAgentsが皮切りとなって、「AIドリブンなDrug Discovery」時代の幕開けとなるのでしょうか。
おわりに
EQUESでは引き続き、「最先端の機械学習技術をあやつり社会の発展を加速する」をミッションに研究開発と社会実装に取り組んでいきます。一緒に事業を創出する仲間を募集しています。詳しくは以下をご覧ください。
Discussion