集団サイズの解析
Stairway Plot 2
Stairway Plot 2
- 特徴: 折り畳まれたSNP頻度スペクトル(SFS)を使用可能。非モデル生物に適用しやすく、過剰適合を抑制。
- 利点: SNPの祖先対立遺伝子の情報が不要、高速計算(前バージョンの10倍以上)、折り畳みSFSでの精度向上。
- 欠点: 情報損失により古代史の推定の不確実性が増す場合がある。
by ChatGPT-4o
SMC++
SMC++の特徴
- 主な機能: SMC++は、個体群の歴史的なサイズ変動を推定するために、部位頻度スペクトル(SFS)と分枝時間分布(TMRCA)を利用します。
- データタイプ: 単一ヌクレオチド多型(SNP)データやハプロタイプデータに対応。
- 計算効率: 高速な計算性能を持ち、大規模なデータセットにも適用可能。
- 結果の解釈: 高解像度の過去の人口動態の変動を推定することができる。
- 必要なデータ量: 高カバレッジのデータが必要であり、広範囲のゲノムデータが有用。
by ChatGPT 4o
MSMC
MSMC(Multiple Sequentially Markovian Coalescent)は、複数の個体のゲノムデータから、共通祖先の時間や人口動態の変化を推定するためのツール
MSMCの特徴
- 主な機能: 複数個体の配列データから、遺伝的共通祖先の時期と人口サイズの変動を推定する。
- データタイプ: 高カバレッジの全ゲノムシーケンスデータ。
- 計算効率: 計算は複雑で時間がかかる場合があるが、複数個体からのデータを同時に扱うことができる。
- 結果の解釈: 高解像度で過去の人口動態を詳細に推定可能。
- 必要なデータ量: 複数個体の高カバレッジデータが必要。
by ChatGPT 4o
MSMC2
MSMC2(Multiple Sequentially Markovian Coalescent 2)は、MSMCの改良版であり、複数のゲノム配列データから共通祖先の時間や人口動態の変動を推定するためのツール
MSMC2の特徴
- 主な機能: 複数個体のゲノムデータから、共通祖先の時間や人口サイズの変動を推定します。MSMCに比べて、より高い解像度で古代の人口動態を推定可能。
- データタイプ: 高カバレッジの全ゲノムシーケンスデータが必要。
- 計算効率: MSMCよりも効率的に動作し、大規模なデータセットにも対応。
- 結果の解釈: MSMC2は、より詳細な人口動態の変動を推定でき、古代の人口分裂や移住パターンの解析が可能。
- 必要なデータ量: 複数個体からの高カバレッジデータが必要。
by Chat GPT 4o
dadi
Dadi-cliは、dadi(Diffusion Approximations for Demographic Inference)ツールのコマンドラインインターフェースであり、部位頻度スペクトル(SFS)を用いて人口動態パラメータを推定するために使用される。
dadi
dadi-cli
dadiの特徴
- 主な機能: 部位頻度スペクトルを用いた人口動態パラメータの推定。
- データタイプ: SNPデータ。
- 計算効率: 高効率で、複雑な人口動態モデルのフィッティングを高速に行える。
- 結果の解釈: 複数の人口動態シナリオをモデル化し、過去の人口サイズ変動、移住率、分岐時期などを推定可能。
- 必要なデータ量: 幅広いゲノムデータに対応可能。
by ChatGPT 4o
森林遺伝育種のデータ解析方法(実践編 5)デモグラフィー推定
PopSizeABC
近似ベイジアン (ABC)を使用した全ゲノムのSNPデータから集団サイズ履歴を推定できるpython and Rスクリプト。
集団サイズの履歴は、多数の集団サイズの変化を含む区分的一定プロセスによってモデル化される。 ABC 推定に使用される要約統計量は、SNP 間の物理的距離の異なるビンにおける対立遺伝子頻度スペクトル (AFS) と平均連鎖不平衡 (LD) 。
Software needs
- Executing python scripts requires a version of python strictly earlier than 3, as well as the libraries numpy and scipy.
- An executable version of ms is provided, but it may not be compatible with all OS. Installing ms and replacing our executable by the new one might be necessary.
- Simulating data with msprime (directory comp_stat.1.zip) instead of ms (directory comp_stat.zip), which we strongly recommend, requires installing msprime following the instructions provided here:
https://pypi.python.org/pypi/msprime - Executing R scripts requires to install the abc library.
Quick start
Genomics, Population Divergence, and Historical Demography of the World's Largest and Endangered Butterfly, The Queen Alexandra's Birdwing
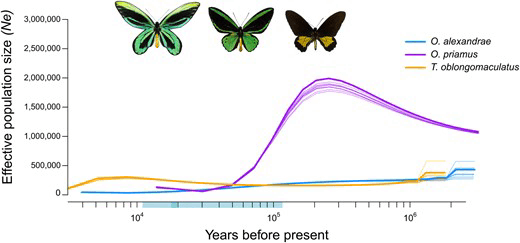
絶滅の危機に瀕しているOrnithoptera alexandraeの2つの個体群の遺伝構造を推定。
ゲノムの多様性や個体群動態について解析。遺伝的多様性が低い結果となったその背景を調べるため、多個体の有効個体群サイズNeの動態を推定できるMSMC2を使用。
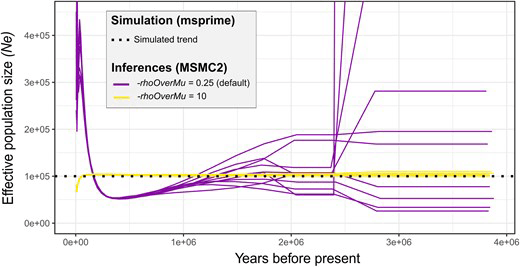
- 理論的研究とシミュレーションにより、SMCダイナミクスは個体群構造と接続性の変化によっても引き起こされる可能性がある(Teixeira et al. 2021)。
- 突然変異率µが組換え率rよりも稀な場合、SMC法はうまく機能しない(Sellinger et al. 2021)。
- 多くの小型非脊椎動物ゲノムの突然変異率µは組み換え率rよりもはるかに低い(Sellinger et al. 2020 ; Sellinger et al. 2021)
- SMCモデルは、組換え率rと突然変異率µの比が1を超えるとうまく機能しない( Sellinger et al. 2021)
シュミレーションデータを使って組み替え率と変異率を調整すると結果が大きく変わったことから、初期調整は重要な役割をもち、SMCの解釈に影響を及ぼす。
実際のデータ
ちょっと小休憩
Fastsimcoal2
fastsimcoal2: demographic inference under complex evolutionary scenarios
fastsimcoal2はfastsimcoalをマルチスレッド化し、集団の近親交配の処理や複雑な人口動態のシナリオの推定を用意にする機能の実装と疎な集団や大きな染色体の効率的なシミュレーションの再実装を実現。
- 事前に指定されたパラメトリックモデルが必要で、パラメトリックアプローチを使用するとノイズの少ない結果が得られる。
Gamma-SMC
Ultrafast genome-wide inference of pairwise coalescence times
各ゲノム位置における2つの相同な染色体のcoalescence timeを推定する超高速なPSMCの再実装ツール。
事後的なcoalescense timeの分布を2つのパラメータを持つガンマ分布として簡潔に表現することで既存の手法よりも10倍以上高速な実装を実現した。
Gamma-SMCは、他のSMCベースの方法と同様に、個体全体に適用するには高品質のPhasingが必要であり、これは小規模データセットでは困難な場合がある。
加えて、Gamma-SMCの精度は既存法と同様ではあるが、推論に遺伝子マップや異なる人口統計モデルを使用したり、事後TMRCA正規化のステップを組み込むこと、推論で対立遺伝子頻度を利用するSFCを使用することで精度がより向上する可能性がある。
ひとまず落ち着いたのでClose