GANSUがRI近似に対応:大規模分子も高速計算可能に、最大21倍の高速化を達成
GPUを用いた量子化学ソフトウェア「GANSU」は、新たに RI近似 (Resolution of the identity approximation) に対応しました。これにより、従来は困難だった100原子を超える大規模分子のHF計算が、高速かつ省メモリで可能になりました。
本記事では、RI近似の簡単な解説とともに、GANSUでの実装戦略、そして PySCF に対して 最大21倍の高速化を達成したベンチマーク結果をご紹介します。
RI近似(密度フィッティング)とは?
量子化学計算における最大のボトルネックの一つが、2電子積分(4中心積分)の計算です。基底関数が
RI近似は、この問題を解決するための近似手法です。RI法では、2電子積分を以下のように2中心と3中心積分で近似します:
ここで
この近似により、メモリ使用量が
GANSUでのRI実装:高速化と省メモリを両立
GANSUでは、以下のような設計でRI法を実装しました:
-
(\mu\nu|P) -
(P|Q)
RI法の導入により、これまで扱えなかった大規模分子にも対応可能となり、計算効率が大きく向上しました。
ベンチマーク結果:最大21倍の高速化、177原子分子で約10倍
以下のグラフは、複数の分子についてRHF計算を実行した際の、SCF収束までの実行時間(秒)を示しています。比較対象として、Pythonベースの量子化学パッケージ PySCFを含めています。
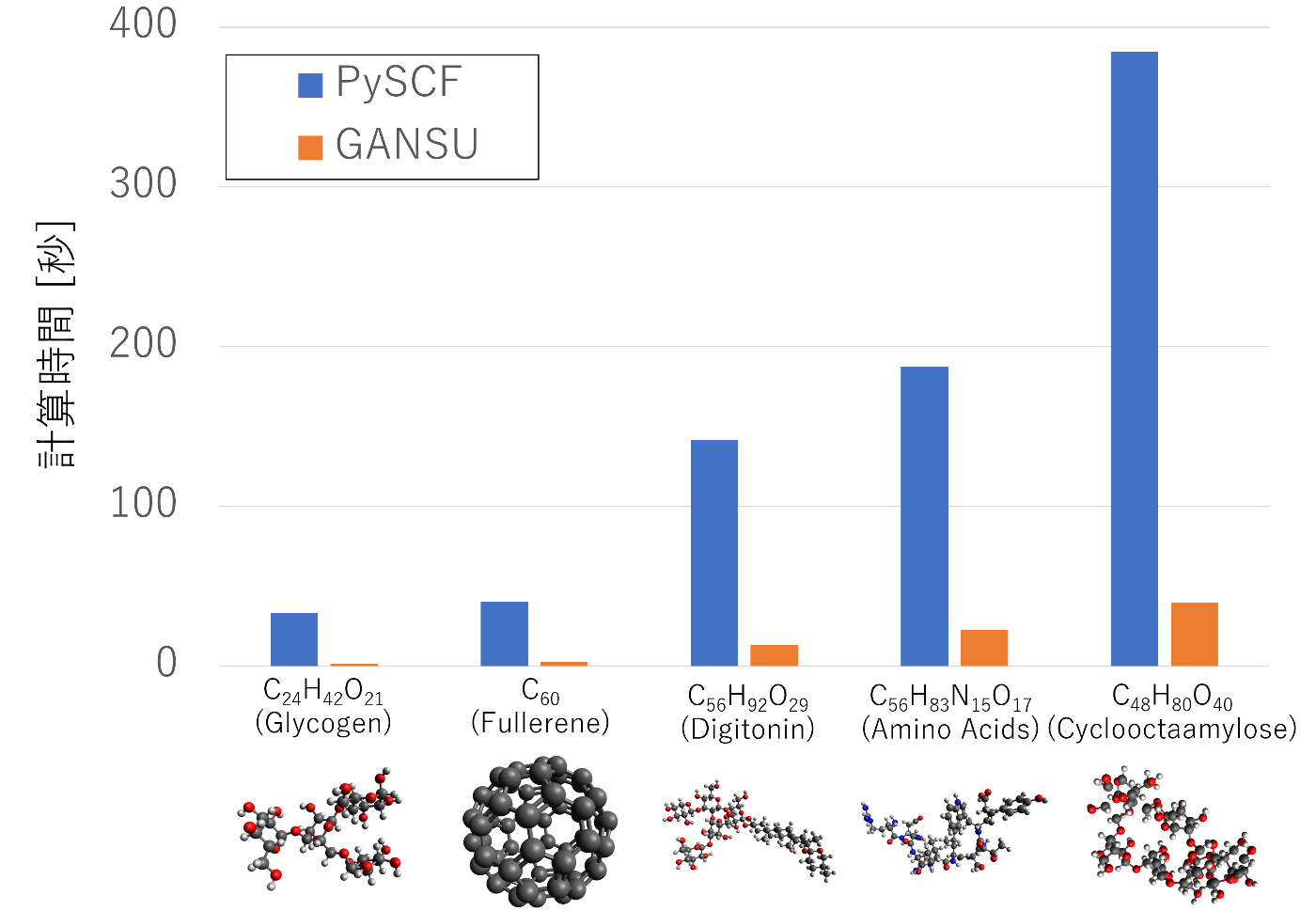
測定環境:NVIDIA A100 + Intel Xeon Gold 6338
使用基底:sto-3g(主基底)、cc-pVDZ-RI(補助基底)
初期値推定:Superposition of Atomic Densities (SAD)
RHF計算、DIIS収束までの時間を測定
このように、最大21倍の高速化を達成しつつ、100原子を超える分子でも1分もかからず計算が可能になりました。
今後の展望
GANSUのRI対応はまだ始まったばかりです。今後は以下のような機能の拡張を予定しています:
- RI-MP2 の実装(ポストHF法への拡張)
- RI-CCSD、RI-CISDなどの実用レベルでのGPU化
まとめ
- GANSUはRI法の導入により、PySCF比で最大21倍の高速化を実現
- 特に大規模分子(177原子)でも約10倍の高速化と10数秒の実行時間を達成
- メモリ使用量も大幅に削減され、現実的なGPUでも実用計算が可能に
- 今後はRI-MP2などポストHF法への展開を予定

広島大学のコンピューティング関連分野で研究を行う研究者が情報を発信します. 主に,量子化学計算や数理最適化に関する技術や研究成果についての記事を掲載しています.
Discussion