もう NCBI GEO の多くの FASTQ を処理しなくてもいいかもしれない #souyakuAC2023
創薬 (dry) Advent Calendar 2023 Day 18 の記事です。 #souyakuAC2023
ケモインフォの記事がほとんどの中、バイオインフォ(遺伝子発現解析)周りの記事です。
本稿では NCBI-generated RNA-seq count data とそのダウンロード補助パッケージ ncbi-counts (PyPI) を紹介します。
GEO とは
Gene Expression Omnibus (GEO) とは、National Center for Biotechnology Information (NCBI) が管理するゲノムデータリポジトリです。研究者が登録した遺伝子発現データなどを取得できます。本稿では NGS で計測したデータについて書きます。
例えば、SARS-CoV-2 の眼球への感染に関するある研究[1] では、眼オルガノイドの single-cell RNA-Seq データ (GSE165477) と角膜・角膜辺縁・強膜の bulk RNA-Seq データ (GSE164073) が登録されています。
後者には以下の 18 サンプルが登録されていて、Title から想像するに、各組織で SARS-CoV-2 感染/非感染 3 サンプルずつの遺伝子発現を計測・登録していそうです。

GSM は 1 つのサンプルに対応し、そのサンプルの条件などが登録されています。GSE は複数の GSM(通常は同一の研究・実験のサンプル)をまとめる単位です。
遺伝子発現量を取得する
サンプルの遺伝子発現量を得るには、通常、2 つの方法があります。
- データ登録者が計算したカウントデータを使う
- Sequence Read Archive (SRA) の生のリードデータを自分で変換する
方法 1 として、ページ下部の Supplementary file にカウントデータが登録してある場合があります。
GSE164073 では GSE164073_Eye_count_matrix.csv.gz が登録されているので、これをダウンロードします。

解凍して GSE164073_Eye_count_matrix.csv の中身を見ると、
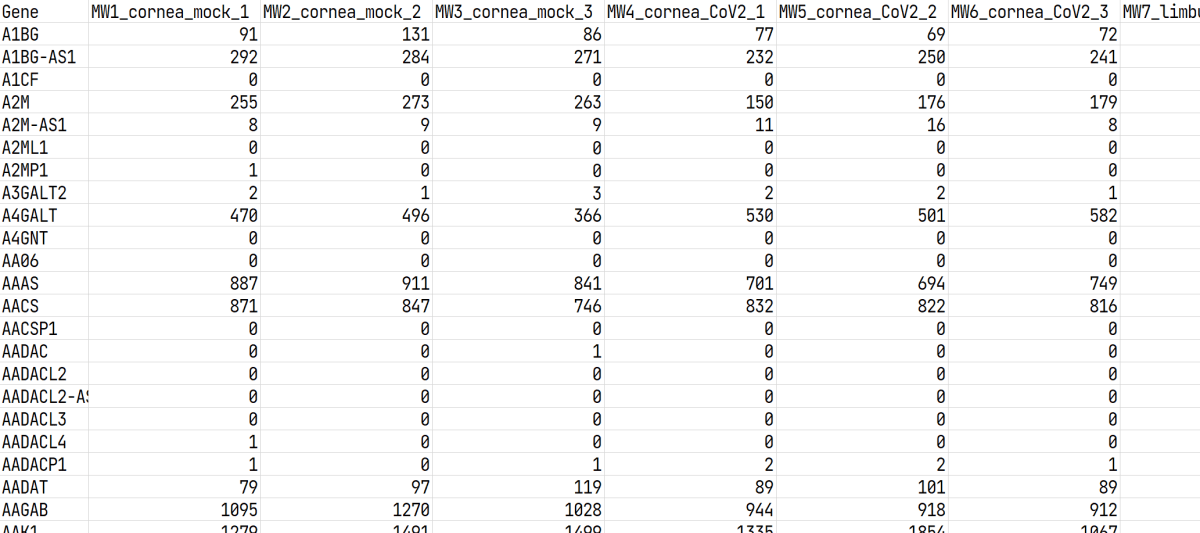
のように、column はサンプル名(18 列)、index は遺伝子シンボル(27,946 行)で、値は対応する発現量になっています。
方法 2 として、Supplementary file の下の SRA Run Selector のリンクから SRA にアクセスします。
GSE164073 では 23.49 Gb 分ののリードデータ(FASTQ ファイル)が登録されているので、これをダウンロードします。

リードデータからカウントデータへの変換方法は多くの解説があるのでここでは説明しません。例えば↓
カウントデータ利用のハードル
ただ、どちらにせよこれらの方法では、下流の解析にすぐ使えるとは限りません。例えば、このようなことが考えられます。
- GEO への登録者がカウントデータをアップロードしていない場合がある。アップロードしてあっても、解析手法やリファレンスファイルやデータ形式が異なったりして、GSE を跨いでの比較可能性に注意が必要
- リードデータからカウントデータへの変換が容易ではない(環境構築がめんどう、ファイルサイズが大きい、計算時間がかかる、など)
NCBI-generated RNA-seq count data とは
これらのハードルを解消すべく、GEO に登録されているリードデータを、NCBI がカウントデータへ変換するという取り組みが始まっていました。X.com でもあまり話題になってなさそうだった[2] ことがこの記事を書く動機でした。

まだ BETA 版ですが、ヒトは計算が終わっていて[3]、今年(2023 年)秋にはマウスについても完了すると記載があります[4]。
NCBI generates RNA-seq count data for only human and mouse RNA-seq runs submitted to GEO. The human RNA-seq count data are currently available and the mouse RNA-seq count data are expected to become available in Fall, 2023.
さて、このカウントデータを取得するには GEO DataSets の検索結果から Download data をクリックするか、https://www.ncbi.nlm.nih.gov/geo/download/?acc={GSExxx} という URL をたたきます。(GSE ページからリンク貼ってくれればいいのに…)
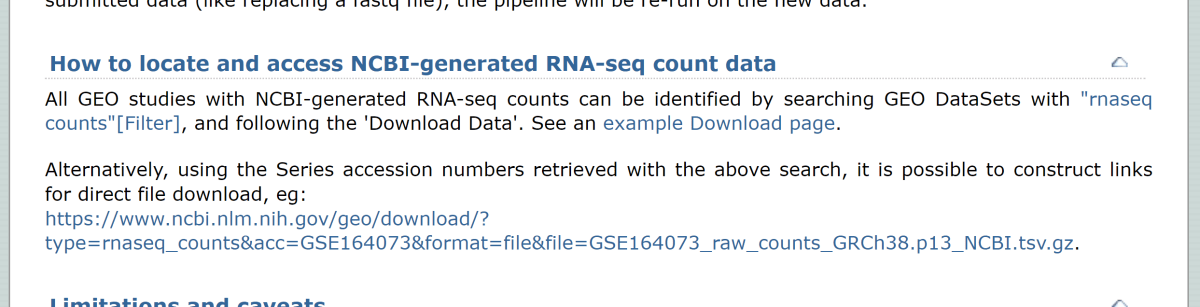
この Download data for GSExxx のページでは、Submitter-supplied data として GSE のページと同じファイルが、そして、NCBI-generated data としてカウントデータ (raw, FPKM, TPM) とアノテーションファイルがあります。

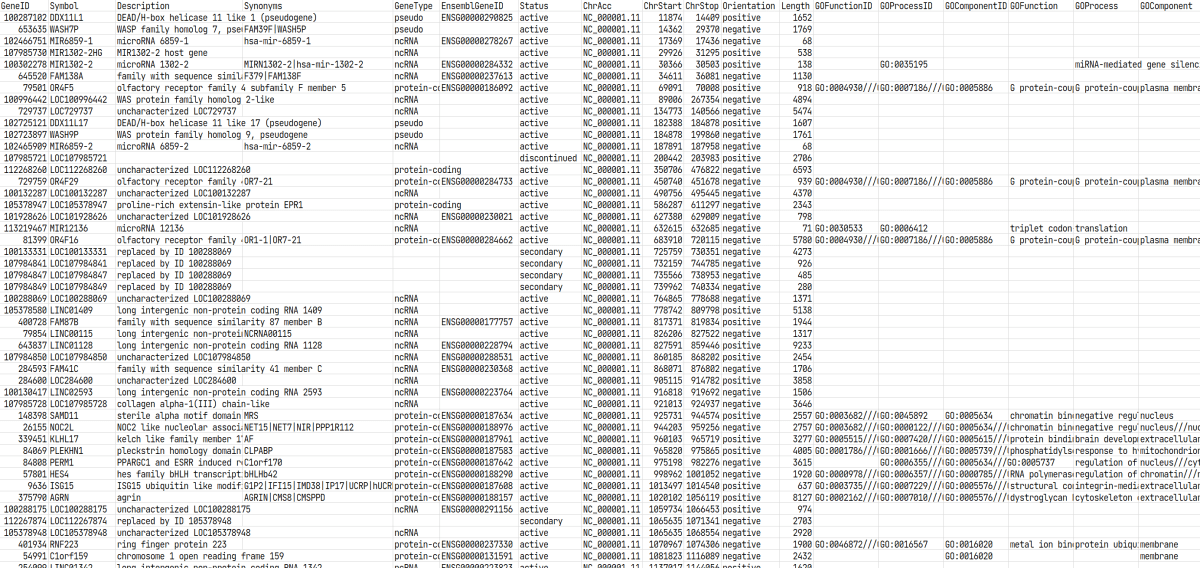
カウントデータのうち、正規化前の発現量の GSE164073_raw_counts_GRCh38.p13_NCBI.tsv は、column がサンプル名(18 列)、index が NCBI Gene ID(39,376 行)です。

アノテーションファイルの Human.GRCh38.p13.annot.tsv は、column がアノテーションの種類(18 列)、index が NCBI Gene ID(39,376 行)です。NCBI Gene ID は一意ですが、HGNC Gene Symbol は一意でなく(このバージョンでは TRNAV-CAC が 3 行存在)、EnsemblGeneID も欠測があります。
ncbi-counts の使い方
ただ、筆者の使い方だとこんな感情が湧いてきました。
- URL の生成がめんどう
- GSE 内の一部のサンプルのみを抽出したい
- カラム名が GSM 番号なので、GSE/GSM のページを見ないとサンプルの属性が分からない
- カウントと(一部の)アノテーションを同じテーブルに結合したい
そこで、これらを解決してカウントデータを便利にダウンロードできる Python パッケージを作ってみました。
- yaml を用意して、コマンドラインで複数のカウントデータを一括ダウンロードする
- GSE 中の欲しい GSM の属性を正規表現で指定する
- アノテーションのうち、欲しいカラムを指定する
ことができます。
例: GSE164073 から各組織のカウントを取得する(コマンドライン)
前述したように、GSE164073 には 3 組織それぞれで mock が 3サンプルと SARS-CoV-2 が 3 サンプルあります。例として、組織ごとのカウントデータを取得してみましょう。まず、このような yaml ファイルを作成して sample_regex.yaml として保存します。
GSE164073:
- control:
title: Cornea
characteristics_ch1: mock
treatment:
title: Cornea
characteristics_ch1: SARS-CoV-2
- control:
title: Limbus
characteristics_ch1: mock
treatment:
title: Limbus
characteristics_ch1: SARS-CoV-2
- control:
title: Sclera
characteristics_ch1: mock
treatment:
title: Sclera
characteristics_ch1: SARS-CoV-2
あとは ncbi-counts をインストールして、yaml ファイルを引数に渡せば実行できます。
-k Symbol はアノテーションのうち、Symbol カラムを残すことを示し、-c は中間ファイルをすべて削除することを示します。他に用意してある引数は PyPI ページの Options を参照。
pip install ncbi-counts
python -m ncbi_counts sample_regex.yaml -k Symbol -c
すると、Cornea のカウントデータが GSE164073-1.tsv として以下のように出力されます。
GeneID 列がユニークな遺伝子 ID で、指定した Symbol カラムに遺伝子シンボルがあり、あとはサンプルごとの発現量です。yaml 中の群 (control, treatment) それぞれの正規表現にマッチしたサンプルについて、群名が GSM の prefix として付与されます。
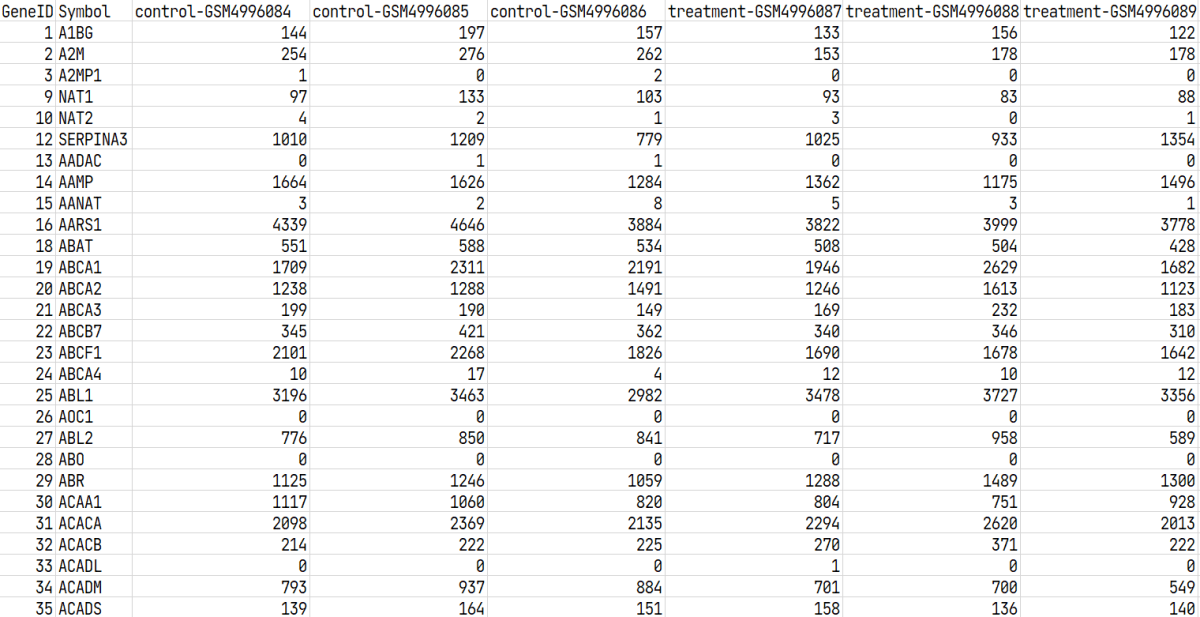
Limbus と Sclera もそれぞれ GSE164073-2.tsv と GSE164073-3.tsv として出力されます。
yaml ファイルの説明
GSE 番号の辞書→出力ファイル内の群名を key とする辞書のリスト→各群サンプルの属性の辞書 の 3 層構造としています。それぞれの辞書の要素は複数でも大丈夫です。
この例では、GSE 番号の辞書は key が GSE164073、出力ファイル内の群名は control, treatment でリストの要素は 3 つ、サンプルの属性の辞書は key が title, characteristics_ch1 で value が対応する正規表現です。
サンプル属性の title は GSM の Title 属性(Cornea_mock_1 や Cornea_CoV2_1)に対応し、characteristics_ch1 は Characteristics 属性(tissue, infection, time point の 3 行)に対応します。


属性の名前には Series の soft ファイルの !Sample_ 以降の文字列を使用できます(GEOparse の仕様)。

ただ、soft ファイルを参照せずとも、SOFT submission instructions ページ内の Sample Attributes や SOFT download を見れば大体対応しそうな属性名は分かると思います。
プログラム中での使用
また、プログラム中で pandas.DataFrame として取得するためには、上の yaml の代わりに同じ構造の辞書を用意することも可能です。
from ncbi_counts import Series
series = Series(
"GSE164073",
[
{
"control": {"title": "Cornea", "characteristics_ch1": "mock"},
"treatment": {"title": "Cornea", "characteristics_ch1": "SARS-CoV-2"},
},
{
"control": {"title": "Limbus", "characteristics_ch1": "mock"},
"treatment": {"title": "Limbus", "characteristics_ch1": "SARS-CoV-2"},
},
# GSM 番号で指定する場合は、例えばこう書けます
{
"control": {"geo_accession": "^GSM499609[6-8]$"},
"treatment": {"geo_accession": "^GSM4996099$|^GSM4996100$|^GSM4996101$"},
},
],
keep_annot=["Symbol"],
save_to=None,
)
series.generate_pair_matrix()
series.cleanup() # コメントアウトすると中間ファイルが残ります
series.pair_count_list[0] # Cornea のカウントデータ(GSE164073-1.tsv と同じ内容)
series.pair_count_list[1] # Limbus のカウントデータ(GSE164073-2.tsv と同じ内容)
series.pair_count_list[2] # Sclera のカウントデータ(GSE164073-3.tsv と同じ内容)
以上が ncbi-counts の大まかな使い方です。
NCBI-generated RNA-seq count data や GEO の構造等について完全に理解しているわけではないので、誤りの指摘・issue・pull request があればお待ちしてます。
-
Eriksen AZ, Møller R, Makovoz B, Uhl SA et al. SARS-CoV-2 infects human adult donor eyes and hESC-derived ocular epithelium. Cell Stem Cell 2021 Jul 1;28(7):1205-1220.e7. ↩︎
-
X 上での
https://www.ncbi.nlm.nih.gov/geo/info/rnaseqcounts.htmlの検索結果は 1 件。 ↩︎ -
ただ、いくつか見てみた感覚では、昔の GSE でも 20-30 % 程度は計算されておらず、何らかの要因で失敗しているものがあるようで、Limitations and caveats の節で言及がされています。失敗した GSE は How NCBI generates RNA-seq count data の節を読んで同じ条件で処理すれば比較可能になるかも? ↩︎
-
ページ自体が "Last modified: April 25, 2023" とのことなので、遅延の可能性も… ↩︎
Discussion